


«Ιϊκή» Γονιδιωματική ("Viral" Genomics): Τίποτα Άλλο Παρά Σειρές Γραμμάτων Σε Μια Τράπεζα Δεδομένων


Μετάφραση: Απολλόδωρος
11 Οκτωβρίου 2021 | ViroLIEgy | Διαβάστε το εδώ
Μπορείτε να κάνετε εφάπαξ ή επαναλαμβανόμενες δωρεές μέσω του Ko-Fi:
«Αν και όλα αυτά είναι καταπληκτικά, λέει ο Calisher, μια σειρά από γράμματα DNA σε μια τράπεζα δεδομένων λέει λίγα ή τίποτα για το πώς πολλαπλασιάζεται ένας ιός, ποια ζώα τον μεταφέρουν, πώς αρρωσταίνει τους ανθρώπους ή αν τα αντισώματα σε άλλους ιούς μπορούν να προστατεύσουν από αυτόν. Απλά μελετώντας τις ακολουθίες, λέει ο Calisher, είναι «σαν να προσπαθείς να πεις αν κάποιος έχει κακή αναπνοή κοιτάζοντας τα δακτυλικά του αποτυπώματα».
Ο ιολόγος Charles Calisher στο Science το 2001
Υπάρχουν πολλά ζητήματα με το να βασιζόμαστε αποκλειστικά στη γονιδιωματική για «ιούς», ειδικά επειδή κανένας «ιός» δεν έχει ποτέ καθαριστεί / απομονωθεί σωστά ούτε έχει αποδειχθεί παθογόνος. Αυτό το βήμα είναι απολύτως απαραίτητο για να γνωρίζουμε εάν αυτές οι σειρές γραμμάτων σε μια βάση δεδομένων υπάρχουν πραγματικά στην πραγματικότητα και έχουν κάποιο νόημα. Όπως προειδοποίησε ο Charles Calisher, η μελέτη των αλληλουχιών DNA δεν λέει τίποτα για έναν «ιό» και χωρίς αυτή την απόδειξη ύπαρξης, τα τυχαία A, C, T, G είναι εντελώς χωρίς νόημα. Παρακάτω παρουσιάζονται τρεις πηγές που υπογραμμίζουν αυτό για το οποίο προειδοποίησε ο Dr. Calisher σχετικά με τις διάφορες προκλήσεις στην προσπάθεια ανάλυσης των «ιογενών» αλληλουχιών και τις δυσκολίες χρήσης της γονιδιωματικής για τον χαρακτηρισμό / ταυτοποίηση των «ιών».
Αυτό το πρώτο άρθρο είναι από τον Ιούλιο του 2016 και καθορίζει τα πολλά ελαττώματα κατά τη χρήση της γονιδιωματικής για «ιογενή» ταυτοποίηση / χαρακτηρισμό. Κάνει εξαιρετική δουλειά περιγράφοντας τις πολυάριθμες προκλήσεις όσον αφορά την ανάλυση των «ιογενών» μεταγονιδιωμάτων. Λάβετε υπόψη ότι με τη μεταγονιδιωματική, δεν υπάρχουν καθόλου προσπάθειες καθαρισμού/απομόνωσης οποιωνδήποτε «ιών». Προσπαθούν να αλληλουχήσουν γονιδιώματα από μικτά δείγματα που περιέχουν ένα πλήθος ξενιστών, βακτηρίων, μυκήτων και DNA "ιού". Είναι αποδεκτό σε αυτό το άρθρο ότι τα θραύσματα από τα "ιϊκά" γονιδιώματα είναι πολύ λιγότερο άφθονα από τις άλλες ουσίες μέσα στο δείγμα. Συζητούν τις διάφορες τεχνολογικές προκλήσεις μαζί με την ποικίλη ποιότητα του λογισμικού και δηλώνουν ότι τα περισσότερα λογισμικά γονιδιωματικής ανάλυσης δεν είναι κατάλληλα για «ιογενή» γονιδιώματα. Αυτές είναι μόνο μερικές από τις πολλές ενδιαφέρουσες αποκαλύψεις μέσα σε αυτή την πηγή:

Προκλήσεις στην ανάλυση των ιϊκών μεταγονιδιωμάτων
«Οι τεχνολογίες αλληλούχισης γονιδιώματος συνεχίζουν να αναπτύσσονται με αξιοσημείωτο ρυθμό, ωστόσο οι αναλυτικές προσεγγίσεις για την ανακατασκευή και ταξινόμηση των ιικών γονιδιωμάτων από μικτά δείγματα παραμένουν περιορισμένες στην απόδοση και τη χρηστικότητα τους. Οι υπάρχουσες λύσεις απευθύνονται γενικά σε έμπειρους χρήστες και συχνά έχουν ασαφές πεδίο εφαρμογής, καθιστώντας δύσκολη την κριτική αξιολόγηση της απόδοσής τους. Υπάρχει αυξανόμενη ανάγκη για διαισθητικά αναλυτικά εργαλεία για ερευνητές που δεν διαθέτουν εξειδικευμένη υπολογιστική εμπειρία και αυτό μπορεί να εφαρμοστεί σε ποικίλες πειραματικές συνθήκες. Αξιοσημείωτες τεχνικές προκλήσεις έχουν εμποδίσει την πρόοδο· για παράδειγμα, τμήματα ιϊκών γονιδιωμάτων είναι συνήθως τάξεις μεγέθους λιγότερο άφθονα από εκείνα των ξενιστών, βακτηρίων και/ή άλλων οργανισμών σε κλινικά και περιβαλλοντικά μεταγονιδιώματα· τα παρατηρούμενα ιϊκά γονιδιώματα συχνά αποκλίνουν σημαντικά από τα γονιδιώματα αναφοράς που απαιτούν τη χρήση εξαντλητικών προσεγγίσεων ευθυγράμμισης· η υψηλή ενδοπληθυσμιακή ιική ποικιλότητα μπορεί να οδηγήσει σε διφορούμενη ανακατασκευή αλληλουχίας· και, τέλος, Τα σχετικά λίγα τεκμηριωμένα ιϊκά γονιδιώματα αναφοράς σε σύγκριση με τον εκτιμώμενο αριθμό διακριτών ιϊκών ταξινομικών κατηγοριών καθιστούν προβληματική την ταξινόμηση. Διάφορα εργαλεία λογισμικού έχουν αναπτυχθεί για να φιλοξενήσουν τις μοναδικές προκλήσεις και περιπτώσεις χρήσης που σχετίζονται με τον χαρακτηρισμό ιϊκών αλληλουχιών. Ωστόσο, η ποιότητα αυτών των εργαλείων ποικίλλει και η χρήση τους συχνά απαιτεί υπολογιστική εμπειρογνωμοσύνη ή πρόσβαση σε ισχυρούς υπολογιστές, περιορίζοντας έτσι τη χρησιμότητά τους σε πολλούς ερευνητές. Σε αυτήν την ανασκόπηση, εξετάζουμε τις γενικές και ειδικές για την εφαρμογή προκλήσεις που θέτει η ιϊκή αλληλουχία και ανάλυση, σκιαγραφούμε το τοπίο των διαθέσιμων εργαλείων και μεθοδολογιών και προτείνουμε τρόπους υπέρβασης των σημερινών εμποδίων στην αποτελεσματική ανάλυση».
«Προς το παρόν έχουμε περιορισμένη αντίληψη της έκτασης της ιϊκής ποικιλότητας που υπάρχει στο περιβάλλον: η έκδοση βάσης δεδομένων του 2014 από τη Διεθνή Επιτροπή για την Ταξινόμηση των Ιών ταξινόμησε μόλις 7 τάξεις, 104 οικογένειες, 505 γένη και 3286 είδη (http://www.ictvonline.org/virustaxonomy.asp). Ωστόσο, μια μελέτη εκτιμά ότι υπάρχουν τουλάχιστον 320.000 είδη ιών που μολύνουν μόνο θηλαστικά (Anthony et al. 2013).
"Για παράδειγμα, ένας ταξινομητής γρήγορης αλληλουχίας μπορεί να αποτύχει εντελώς να ανιχνεύσει ένα νέο στέλεχος ενός καλά χαρακτηρισμένου ιού και εξίσου μπορεί να αποδώσει καλά με τις ακολουθίες Illumina, αλλά να προσφέρει φτωχά αποτελέσματα για δεδομένα που παράγονται με την πλατφόρμα Ion Torrent. Επιπλέον, τα αποτελέσματα που προκύπτουν από αυτές τις αναλύσεις θα πρέπει να είναι αναπαραγώγιμα, κατανοητά και χρήσιμα για τον τελικό χρήστη, με πρόβλεψη για ποιοτικό έλεγχο και διαχείριση σφαλμάτων.
«Μεθοδολογικά, τα περισσότερα λογισμικά ανάλυσης γονιδιωματικής αλληλουχίας δεν είναι κατάλληλα για ιικά γονιδιώματα. Τα γενικά εργαλεία που είναι σε θέση να αντιμετωπίσουν τις προκλήσεις που θέτουν οι ιικές αλληλουχίες είναι συχνά εφαρμόσιμα μόνο σε περιορισμένες κυκλοφορίες. Η επιλογή μεταξύ προσεγγίσεων καθίσταται δύσκολη λόγω της αφθονίας ανόμοιων αλλά λειτουργικά ισοδύναμων μεθοδολογιών και, γενικά, της έλλειψης αυστηρών σημείων αναφοράς για τα ιικά σύνολα δεδομένων. Ενώ υπάρχει πολλή συνεχιζόμενη έρευνα σε αυτόν τον τομέα, τόσο η ευαίσθητη ανίχνευση προηγουμένως χαρακτηρισμένων ιών όσο και η ανακάλυψη ιών παραμένουν βασικές προκλήσεις ανοιχτές για καινοτομία».
2. Εμπλουτισμός ιϊκής αλληλουχίας: φυσικές και in silico προσεγγίσεις
«Εντός των μεταγονιδιωμάτων η αναλογία των ιικών νουκλεϊνικών οξέων είναι συνήθως πολύ χαμηλότερη από εκείνη του ξενιστή ή άλλων μικροβίων, περιορίζοντας την ποσότητα του σήματος που είναι διαθέσιμο για ανάλυση μετά την αλληλούχιση. Για να μετριαστεί αυτό το ζήτημα, χρησιμοποιούνται ευρέως προσεγγίσεις εμπλουτισμού και ενίσχυσης πριν από την αλληλούχιση ιικών δειγμάτων. Η διήθηση μεγέθους ή ο εμπλουτισμός με βάση την πυκνότηταμε φυγοκέντρηση είναι δύο αποτελεσματικές μέθοδοι για την αύξηση της απόδοσης του ιού, αν και τέτοιες μέθοδοι μπορεί να επηρεάσουν την παρατηρούμενη σύνθεση των ιικών πληθυσμών (Ruby, Bellare και Derisi 2013). Εναλλακτικά, η ενίσχυση PCR μπορεί να χρησιμοποιηθεί για τη δημιουργία αφθονίας συγκεκριμένων ιικών αλληλουχιών που υπάρχουν σε ένα δείγμα, μια ευρέως χρησιμοποιούμενη στρατηγική, η οποία χρησιμοποιήθηκε στην ταυτοποίηση και ανάλυση του κορονοϊού MERS (Zaki et al. 2012; Cotten et al. 2013, 2014), αν και ο αποτελεσματικός σχεδιασμός εκκινητών μπορεί να είναι δύσκολος παρουσία υψηλής γονιδιωματικής ποικιλότητας στα ιικά είδη-στόχους. Αντίθετα, μια υπερβολική κάλυψη αλληλούχισης μπορεί να οδηγήσει στην κατασκευή υπερβολικά πολύπλοκων και δυσκίνητων de novo γραφημάτων συναρμολόγησης παρουσία υψηλής γονιδιωματικής ποικιλομορφίας, μειώνοντας την ποιότητα συναρμολόγησης. Χρησιμοποιώντας in silico κανονικοποίηση (Crusoe et al. 2015), η πλεονάζουσα κάλυψη μπορεί να μειωθεί με την απόρριψη αλληλουχιών που περιέχουν περιττές πληροφορίες. Αυτή η προσέγγιση αυξάνει την αναλυτική αποτελεσματικότητα όταν ασχολούμαστε με δεδομένα αλληλουχίας υψηλής κάλυψης και έχουμε δείξει ότι μπορεί να ωφελήσει την de novo συναρμολόγηση αλληλουχιών ιϊκής συναίνεσης. Μια άλλη στρατηγική in silico για την αύξηση της αναλυτικής αποτελεσματικότητας με την απόρριψη περιττών δεδομένων είναι το φιλτράρισμα αλληλουχιών από γνωστούς άφθονους οργανισμούς μέσω ευθυγράμμισης με ένα ή περισσότερα γονιδιώματα αναφοράς χρησιμοποιώντας έναν νάρθηκα ή ένα εξειδικευμένο εργαλείο (προσεγγίσεις που εξετάστηκαν στο Daly et al. 2015).
4. Συναρμολόγηση γονιδιωμάτων: denovo και συναρμολόγηση βάσει αναφοράς
"Η ανακατασκευή της αλληλούχισης σε γονίδια πλήρους μήκους και γονιδιώματα μπορεί να πραγματοποιηθεί είτε μέσω ευθυγράμμισης βάσει αναφοράς είτε μέσω de novo συναρμολόγησης, μια απόφαση που εξαρτάται από πειραματικούς στόχους, μήκος ανάγνωσης, ποιότητα και πολυπλοκότητα δεδομένων. Σε προσεγγίσεις που βασίζονται σε αναφορές, οι αναγνώσεις χαρτογραφούνται σε παρόμοιες περιοχές ενός παρεχόμενου γονιδιώματος προτύπου, μια καλά μελετημένη και υπολογιστικά αποτελεσματική διαδικασία που υλοποιείται με έναν δείκτη πίνακα επιθημάτων του γονιδιώματος αναφοράς. Αντίθετα, η de novo συναρμολόγηση είναι υπολογιστικά εξαντλητική, αλλά σημαντική σε περιπτώσεις όπουείτε ένα γονιδίωμα-στόχος δεν χαρακτηρίζεται επαρκώς είτε επιδιώκεται η ανακατασκευή γονιδιωμάτων a priori άγνωστων οντοτήτων σε μεταγονιδιώματα, όπως σε μελέτες επιτήρησης. Για δεδομένα σύντομης ανάγνωσης, το αυξημένο μήκος αλληλουχίας που παρέχεται από τη συναρμολόγηση μπορεί να είναι απαραίτητο για τη διάκριση των μελών των εξαιρετικά διατηρημένων οικογενειών γονιδίων μεταξύ τους. Η συναρμολόγηση χρησιμοποιείται επίσης ευρέως για τη δημιουργία αλληλουχιών συναίνεσης ολόκληρου του γονιδιώματος για τη διευκόλυνση των αναλύσεων της ιικής παραλλαγής και είναι ένα τυπικό σημείο εκκίνησης για αναλύσεις διαφορετικών πληθυσμών καλά χαρακτηρισμένων ιών. Ακόμη και όταν υπάρχουν διαθέσιμες μεγάλες αναγνώσεις, η συναρμολόγηση παίζει σημαντικό ρόλο στον μετριασμό των υψηλών ποσοστών σφάλματος που σχετίζονται με τις τεχνολογίες αλληλούχισης ενός μορίου, αποδίδοντας ακριβείς αλληλουχίες συναίνεσης από ανακριβείς μεμονωμένες αναγνώσεις.
4.2 Συναρμολόγηση Denovo για μεταγονιδιώματα
«Οι τυπικοί de novo συναρμολογητές έχουν σχεδιαστεί για να ανακατασκευάζουν γονιδιώματα με ομοιόμορφη κάλυψη αλληλούχισης σε όλο το μήκος τους. Αυτό είναι προβληματικό για τα μεταγονιδιώματα (συμπεριλαμβανομένων των viromes) όπου η κάλυψη συνήθως ποικίλλει σημαντικά τόσο μεταξύ διαφορετικών γονιδιωμάτων όσο και εντός μεμονωμένων γονιδιωμάτων.
"Αν και οι συναρμολογητές μεταγονιδιώματος γενικά ξεπερνούν τους συναρμολογητές ενός γονιδιώματος στην ανακατασκευή διαφορετικών γονιδιωμάτων ταυτόχρονα, η πολυπλοκότητα αυτού του έργου καθορίζει την τάση τους να καταρρέουν την παραλλαγή στο ή κάτω από το στέλεχος level σε αλληλουχίες συναίνεσης. Ακόμη και για το σκοπό αυτό, η αποτελεσματικότητά τους μπορεί να είναι περιορισμένη ως συνέπεια της ακραίας διακύμανσης εντός συγκεκριμένων πληθυσμών ιών RNA λόγω μετάλλαξης και ανασυνδυασμού και της χαμηλής ή/και άνισης κάλυψης αλληλούχισης σε ένα συγκεκριμένο γονιδίωμα. Επιπλέον, πρέπει να σημειωθεί ότι η de novo συναρμολόγηση είναι ιδιαίτερα ευαίσθητη στην ποιότητα των αλληλουχιών εισόδου, πράγμα που σημαίνει ότι τα προβλήματα κατά την εξαγωγή δειγμάτων, τον εμπλουτισμό και την προετοιμασία της βιβλιοθήκης μπορεί να είναι ιδιαίτερα επιζήμια για τις κατάντη αναλύσεις. Ως εκ τούτου, καίριας σημασίας είναι οι μέθοδοι ποιοτικού ελέγχου για την ανίχνευση και, κατά περίπτωση, τη διόρθωση προβλημάτων που σχετίζονται με τη μόλυνση (Darling et al. 2014; Orton et al. 2015), ανάγνωση εκκινητών και αναγνώσεις χαμηλής ποιότητας (αναθεωρήθηκε στο Leggett et al. 2013)."
5. Ανακατασκευή απλότυπου σε συγκεκριμένους ιϊκούς πληθυσμούς
«Τα ιϊκά γονιδιώματα και τα μεταγονιδιώματα που περιλαμβάνουν υψηλή ενδοειδική παραλλαγή μπορεί να είναι δύσκολοι στόχοι για συναρμολόγηση, δημιουργώντας πολύπλοκα γραφήματα συναρμολόγησης και κατακερματισμένα συγκροτήματα. Αυτό συμβαίνει συχνά για κλινικά δείγματα από ασθενείς με HIV και ηπατίτιδα C, στα οποία τα υψηλά ποσοστά μετάλλαξης και οι μεγάλες διάρκειες μόλυνσης μπορούν να συμβάλουν στην ακραία απόκλιση του πληθυσμού, αλλά μπορούν επίσης να παρατηρηθούν σε περιβαλλοντικά δείγματα.
"Ένας περιορισμός όλων αυτών των προσεγγίσεων. Ωστόσο, είναι η εξάρτησή τους από μια ενιαία αλληλουχία αναφοράς με την οποία θα εκτελεστεί η αρχική ευθυγράμμιση, μια διαδικασία που προϋποθέτει ένα βαθμό ομοιότητας αλληλουχίας που μπορεί να μην παρατηρείται πάντα σε διάφορες περιοχές, όπως περιοχές που κωδικοποιούν πρωτεΐνες φακέλου, γονιδιωμάτων ιών RNA.
6. Ταξινόμηση ακολουθίας
"Η ταξινόμηση αλληλουχιών είναι ένα από τα πιο μελετημένα προβλήματα στην υπολογιστική βιολογία και η ταξινομική ανάθεση αποτελεί βασικό στόχο της ανάλυσης μεταγονιδιώματος. Όλες οι μέθοδοι ταξινόμησης, σε κάποιο βαθμό, εξαρτώνται από την ανίχνευση ομοιότητας μεταξύ μιας ακολουθίας ερωτημάτων και μιας συλλογής σχολιασμένων ακολουθιών. Η ταξινόμηση μπορεί να πραγματοποιείται είτε με τη χρήση μη συναρμολογημένων ενδείξεων είτε με ανακατασκευασμένα στοιχεία που προκύπτουν από τη διαδικασία συναρμολόγησης. Οι υπολογιστικές απαιτήσεις των διαθέσιμων προσεγγίσεων ποικίλλουν δραματικά ανάλογα με την ικανότητά τους να ανιχνεύουν ομολογία σε αποκλίνουσες αλληλουχίες.
6.1. Αναζητήσεις ομοιότητας αλληλουχίας
"Οι προσεγγίσεις αναγνώρισης ιών εξαρτώνται συνήθως από αναζητήσεις ομοιότητας σε μια βάση δεδομένων χρησιμοποιώντας έναν νάρθηκα όπως το BLAST (Altschul et al. 1990). Μπορούν να χρησιμοποιηθούν ολοκληρωμένες βάσεις δεδομένων (π.χ. GenBank) ή μικρότερες προσαρμοσμένες βάσεις δεδομένων που περιέχουν, για παράδειγμα, μόνο ιογενείς αλληλουχίες ενδιαφέροντος, αν και οι τελευταίες μπορούν να παράγουν παραπλανητικά αποτελέσματα.
6.2 Εναλλακτικές λύσεις στις αναζητήσεις ομοιότητας
"Αν και οι εξαντλητικές μέθοδοι τύπου BLAST μπορούν να ανιχνεύσουν ομολογία σε αποκλίνουσες αλληλουχίες, αυτές οι μέθοδοι περιορίζονται γενικά από τις σχετικά λίγες επικυρωμένες ιικές αλληλουχίες που κατατίθενται σε δημόσιες βάσεις δεδομένων, την υψηλή ποικιλομορφία εντός των ιογενών οικογενειών που μπορεί να αποκρύψει τη συγγένεια και την έλλειψη ενός καθορισμένου συνόλου βασικών γονιδίων κοινών σε όλους τους ιούς που μπορούν να χρησιμοποιηθούν για τη διάκριση ειδών (π.χ. το γονίδιο 16S για βακτήρια) (Fancello, Raoult, και Desnues 2012). Αυτά τα χαρακτηριστικά καθιστούν δύσκολη την εκχώρηση ορίων ομοιότητας για ταξινόμηση που ισχύουν για όλους τους πιθανούς ιούς σε ένα δείγμα (Simmonds 2015).
«Μια θεμελιώδης πρόκληση στην ταξινόμηση των ιικών αλληλουχιών με οποιαδήποτε από αυτές τις μεθόδους παραμένει η περιορισμένη εκπροσώπησή τους σε επιμελημένες βάσεις δεδομένων αλληλουχίας. Ενώ ο ρυθμός με τον οποίο προστίθενται νέοι ιοί στη συλλογή RefSeq του NCBI έχει αυξηθεί σημαντικά, από μέσο όρο έτους 0,34 είδη/ημέρα το 2010 σε 2,5 είδη/ημέρα το 2015 (Σχήμα 3), η τεκμηριωμένη κατανόησή μας για την έκταση της ιικής διαφοράςπαραμένει επιφανειακή (Anthony et al. 2013). Επομένως, οι αναγνώσεις της πραγματικής ιικής προέλευσης κινδυνεύουν να χαθούν σε πολλές περιπτώσεις. Ο ρυθμός αύξησης των βάσεων δεδομένων υπογραμμίζει επίσης την ανάγκη διατήρησης συχνά ενημερωμένων ευρετηρίων αναζήτησης για την ταξινόμηση ακολουθιών, η κατασκευή των οποίων συχνά απαιτεί εξειδικευμένους διακομιστές εξοπλισμένους με εκατοντάδες gigabytes μνήμης RAM. Ακόμη και αν διατηρούνται ενημερωμένα ευρετήρια μέσα σε ένα δημόσιο αποθετήριο, τα μεγέθη των αρχείων τους είναι σημαντικά, απαιτώντας από τους χρήστες να έχουν πρόσβαση σε γρήγορη σύνδεση στο διαδίκτυο. Κατά συνέπεια, η πλήρης εξωτερική ανάθεση της ταξινόμησης αλληλουχιών σε απομακρυσμένες υπηρεσίες ιστού είναι μια συναρπαστική προοπτική για όσους έχουν επαρκείς συνδέσεις στο διαδίκτυο αλλά χωρίς ισχυρό υπολογιστικό υλικό, αυξάνοντας το πεδίο για τη διεξαγωγή αναλύσεων με φορητούς υπολογιστές.
"Επιπλέον, η ταξινόμηση των ιικών αλληλουχιών εξαρτάται σε μεγάλο βαθμό από την ποιότητα των επιμελημένων ιογενών βάσεων δεδομένων όπως η RefSeq, στις οποίες η υποβολή πρόσφατα ανακαλυφθεισών αλληλουχιών μπορεί να είναι απαγορευτικά χρονοβόρα".

Αυτό το δεύτερο άρθρο είναι από τον Ιανουάριο του 2017. Αναλύει τις τρεις κύριες μεθόδους που χρησιμοποιούνται για την αλληλουχία των "ιών" και παρέχει τα πλεονεκτήματα και τα μειονεκτήματα του καθενός. Λάβετε υπόψη κατά την ανάγνωση αυτού, το αρχικό γονιδίωμα "SARS-COV-2" προήλθε από τη μεταγονιδιωματική ανάλυση μη καθαρισμένου/μη απομονωμένου BALF που ελήφθη από έναν ασθενή. Ανέλυσα λεπτομερώς τη διαδικασία εδώ:
Κατασκευάζοντας το γονιδίωμα του "SARS-COV-2"

Οι τρεις κύριες μέθοδοι που περιγράφονται στο άρθρο περιλαμβάνουν τη μεταγονιδιωματική, την ενίσχυση PCR και τον εμπλουτισμό στόχου. Ανεξάρτητα από το τι υποστηρίζεται, καμία από αυτές τις μεθόδους δεν είναι κατάλληλη για την ανίχνευση νέων «ιών», ειδικά όταν αλληλουχούνται από πηγές που δεν έχουν περάσει από κατάλληλα στάδια καθαρισμού (φυγοκέντρηση, διήθηση, καθίζηση κ.λπ.) για την απομάκρυνση του DNA του ξενιστή, των βακτηρίων, των μυκήτων και άλλων πιθανών ξένων μικροοργανισμών / ρύπων από το αρχικό δείγμα. Όπως θα δείτε, είτε το δείγμα περιέχει κάτι περισσότερο από το στοχευμένο "ιογενές" υλικό είτε η αλληλουχία πρέπει να είναι ήδη γνωστή εκ των προτέρων.
Κλινικές και βιολογικές γνώσεις από την αλληλούχιση του ιικού γονιδιώματος
«Η αλληλούχιση των ιϊκών νουκλεϊκών οξέων, είτε από καλλιέργειες είτε απευθείας από κλινικά δείγματα, περιπλέκεται από την παρουσία μολυσμένου DNA ξενιστή58. Αντίθετα, το μεγαλύτερο μέρος της βακτηριακής αλληλουχίας πραγματοποιείται επί του παρόντος σε κλινικές απομονώσεις που καλλιεργούνται. Έτσι, η προετοιμασία του δείγματος είναι σχετικά απλή (Πίνακας 2 και ανασκόπηση στο Ref. 59). Επί του παρόντος, η αλληλούχιση του γονιδιώματος των ιών μπορεί να επιτευχθεί με εξαιρετικά βαθιά αλληλούχιση ή μέσω του εμπλουτισμού για ιικά νουκλεϊκά οξέα πριν από την αλληλούχιση, είτε άμεσα είτε με συγκέντρωση σωματιδίων ιού. Όλες αυτές οι προσεγγίσεις έχουν το δικό τους κόστος και πολυπλοκότητα.
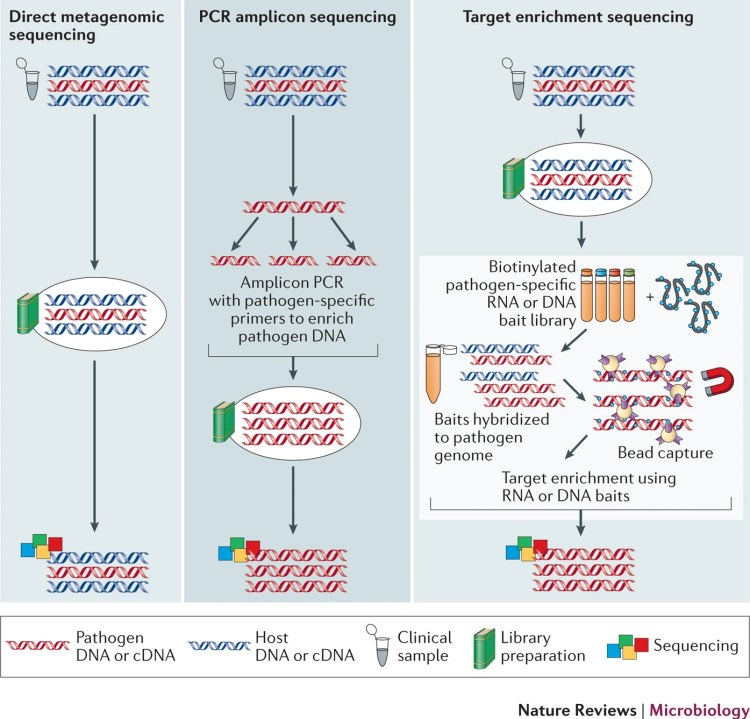
Τρεις κύριες μέθοδοι χρησιμοποιούνται επί του παρόντος για την αλληλούχιση του ιϊκού γονιδιώματος: μεταγονιδιωματική αλληλούχιση, αλληλούχιση αμπλικονίου PCR και αλληλούχιση εμπλουτισμού στόχου (Σχήμα 1).
Μεταγονιδιωματική. Οι μεταγονιδιωματικές προσεγγίσεις έχουν χρησιμοποιηθεί εκτενώς για την ανακάλυψη παθογόνων και για τον χαρακτηρισμό της μικροβιακής ποικιλότητας σε περιβαλλοντικά και κλινικά δείγματα60,61. Το συνολικό DNA και/ή RNA, συμπεριλαμβανομένων των βακτηρίων, των ιών, των μυκήτων και άλλων παθογόνων παραγόντων, εξάγονται από ένα δείγμα και μια βιβλιοθήκη προετοιμάζεται και αλληλουχείται με αλληλούχιση κυνηγετικού όπλου ή αλληλούχιση RNA (RNA-seq). Στο πλαίσιο 1 διερευνώνται οι διαγνωστικές εφαρμογές για τη μεταγονιδιωματική και το RNA-seq· Για παράδειγμα, σε εγκεφαλίτιδα άγνωστης αιτιολογίας62,63,64, για τις οποίες οι συμβατικές μέθοδοι όπως η PCR συχνά δεν είναι διαγνωστικές, η μεταγονιδιωματική και το RNA-seq έχουν ανιχνεύσει ιογενείς λοιμώξεις65,66,67 και άλλες αιτίες68 εγκεφαλίτιδας. Επιπλέον, αυτές οι μέθοδοι έχουν χρησιμοποιηθεί για την αλληλουχία ολόκληρου του γονιδιώματος ορισμένων ιών, συμπεριλαμβανομένου του ιού Epstein-Barr (EBV)69 και HCV27. Ωστόσο, σε κλινικά δείγματα, η παρουσία μολυσματικών νουκλεϊνικών οξέων από τον ξενιστή και τους κοινούς μικροοργανισμούς58 (Πίνακας 2) μειώνει την ευαισθησία. Το ποσοστό των αναγνώσεων που ταιριάζουν με το γονιδίωμα του ιού-στόχου από το μεταγονιδιωματικό WGS είναι συχνά χαμηλό. για παράδειγμα, 0,008% για EBV στο αίμα ενός υγιούς ενήλικα70, 0,0003% για τον ιό Lassa σε κλινικά δείγματα71 και 0,3% για τον ιό Zika σε δείγμα που εμπλουτίστηκε για σωματίδια ιού μέσω διήθησης και φυγοκέντρησης72. Το βάθος ανάγνωσης είναι συχνά ανεπαρκές για την ανίχνευση αντίστασης27 και το κόστος είναι υψηλό. Έτσι, η μεταγονιδιωματική αλληλουχία έχει συνήθως πραγματοποιηθεί μόνο σε μικρό αριθμό δειγμάτων για ερευνητικούς σκοπούς72,73. Η συγκέντρωση των σωματιδίων του ιού (δείτε το παράδειγμα του ιού Zika παραπάνω72), η εξάντληση του υλικού ξενιστή ή/και η αλληλούχιση σε υψηλό βάθος ανάγνωσης μπορεί να αυξήσει την ποσότητα της αλληλουχίας ιών, αλλά όλες αυτές οι μέθοδοι αυξάνουν το κόστος. Η συγκέντρωση σωματιδίων ιού από κλινικά δείγματα με αναρρόφηση μέσω αντισωμάτων (για παράδειγμα, ανακάλυψη ιού βασισμένη σε πολυμορφισμό μήκους θραυσμάτων ενισχυμένου με συμπληρωματικό DNA (cDNA) (AFLP), συντομογραφία VIDISCA), διήθηση, υπερφυγοκέντρηση και εξάντληση των ελεύθερων νουκλεϊνικών οξέων, τα οποία προέρχονται κυρίως από τον ξενιστή, έχουν όλα δοκιμαστεί74,75,76,77Ωστόσο, αυτές οι μέθοδοι μπορούν επίσης να μειώσουν τη συνολική ποσότητα ιικών νουκλεϊνικών οξέων, έτσι ώστε να είναι ανεπαρκής για την προετοιμασία μιας βιβλιοθήκης αλληλούχισης. Μη ειδικές μέθοδοι ενίσχυσης, όπως η ενίσχυση πολλαπλής μετατόπισης (MDA), οι οποίες χρησιμοποιούν τυχαίους εκκινητές και πολυμεράσες Φ29, μπορούν να αυξήσουν την απόδοση του DNA. Ωστόσο, αυτές οι προσεγγίσεις είναι χρονοβόρες, δαπανηρές και μπορεί να αυξήσουν τον κίνδυνο προκαταλήψεων, σφαλμάτων και μόλυνσης, χωρίς απαραίτητα να βελτιώνουν την ευαισθησία78,79. Επιπλέον, το ποσοστό των αναγνώσεων υποδοχής παραμένει συχνά υψηλό80.
Όταν χρησιμοποιούνται μεταγονιδιωματικές μέθοδοι για την ανακάλυψη ή τη διάγνωση παθογόνων, είναι ζωτικής σημασίας να χρησιμοποιούνται κατάλληλα εργαλεία βιοπληροφορικής και βάσεις δεδομένων που μπορούν να αξιολογήσουν εάν οι ανιχνευόμενες αλληλουχίες παθογόνων είναι πιθανό να είναι η αιτία μόλυνσης, τυχαίων ευρημάτων ή μολυσματικών παραγόντων. Οι βιοπληροφορικές αναλύσεις μεγάλων συνόλων μεταγονιδιωματικών δεδομένων απαιτούν υπολογιστικούς πόρους υψηλής απόδοσης».
Ωστόσο, τυχαία ευρήματα, τόσο σε αλληλουχίες ξενιστών όσο και μικροβίων, μπορεί επίσης να παρουσιάσουν ηθικά και ακόμη και διαγνωστικά διλήμματα για την κλινική μεταγονιδιωματική82 (βλ. παρακάτω). Ένα πρόσφατο παράδειγμα αφορούσε ένα σύμπλεγμα περιπτώσεων οξείας χαλαρής μυελίτιδας που σχετίζονταν με τον εντεροϊό D68 (Σχετ. 83). Τα μεταγονιδιωματικά δεδομένα από δείγματα που ελήφθησαν από ασθενείς έδειξαν την παρουσία εναλλακτικών παθογόνων, μερικά από τα οποία είναι θεραπεύσιμα, και συζητήθηκαν επίσημα84 και ανεπίσημα επιστημονικά κανάλια (βλ. άρθρο Omicsomics blogspot)."
"Εμπλουτισμός PCR amplicon. Μια εναλλακτική λύση στις μεταγονιδιωματικές προσεγγίσεις είναι ο εμπλουτισμός του συγκεκριμένου ιικού γονιδιώματος πριν από την αλληλούχιση. Η ενίσχυση PCR του ιικού γενετικού υλικού χρησιμοποιώντας εκκινητές που είναι συμπληρωματικοί σε μια γνωστή αλληλουχία νουκλεοτιδίων ήταν η πιο κοινή προσέγγιση για τον εμπλουτισμό μικρών ιικών γονιδιωμάτων, όπως ο ιός HIV και ο ιός της γρίπης.
«Αλληλεπικαλυπτόμενα PCR σε συνδυασμό με NGS έχουν χρησιμοποιηθεί για την αλληλουχία ολόκληρου του γονιδιώματος μεγαλύτερων ιών, όπως ο HCMV93, αλλά αυτή η μέθοδος έχει περιορισμένη επεκτασιμότητα, καθώς απαιτούνται πολλοί εκκινητές και σχετικά μεγάλη ποσότητα αρχικού DNA93. Αυτό περιορίζει τον αριθμό των κατάλληλων δειγμάτων που είναι διαθέσιμα και επίσης τα γονιδιώματα που μπορούν να μελετηθούν χρησιμοποιώντας αυτή τη μέθοδο. Για παράδειγμα, απαιτήθηκαν 8-19 προϊόντα PCR για την ενίσχυση του γονιδιώματος του ιού Έμπολα39, και δύο μελέτες νοροϊού χρειάστηκαν 14 και 22 προϊόντα PCR, αντίστοιχα86,87. Για κλινικές εφαρμογές αυτό είναι προβληματικό λόγω του υψηλού εργαστηριακού φόρτου εργασίας που σχετίζεται με πολυάριθμες διακριτές αντιδράσεις PCR, της ανάγκης για ατομική ομαλοποίηση των συγκεντρώσεων διαφορετικών amplicons PCR πριν από τη συγκέντρωση, της αυξανόμενης πιθανότητας αποτυχίας αντίδρασης λόγω αναντιστοιχίας εκκινητών (ιδιαίτερα για εξαιρετικά μεταβλητούς ιούς) και του υψηλού κόστους εργασίας και αναλωσίμων94. Επομένως, παρόλο που η αλληλούχιση ιών με βάση PCR τόσο μεγάλη όσο 250 kb είναι τεχνικά εφικτή, η αναλογική σχέση μεταξύ του μεγέθους του γονιδιώματος και της τεχνικής πολυπλοκότητας καθιστά την αλληλούχιση με βάση την PCR ιικών γονιδιωμάτων που είναι πάνω από 20-50 kb ανέφικτη με τις τρέχουσες τεχνολογίες, ιδιαίτερα για μεγάλες μελέτες πολλαπλών δειγμάτων ή διαγνωστικά ρουτίνας. Μια άλλη σκέψη είναι ότι ο αυξανόμενος αριθμός αντιδράσεων PCR απαιτεί αντίστοιχη αύξηση της ποσότητας του δείγματος και αυτό δεν είναι πάντα δυνατό καθώς τα κλινικά δείγματα είναι περιορισμένα».
«Εξαιρετικά μεταβλητά παθογόνα, ιδιαίτερα εκείνα που έχουν ευρέως αποκλίνουσες γενετικές γενεαλογίες ή γονότυπους, όπως ο HCV97 και ο νοροϊός, προκαλούν προβλήματα στην ενίσχυση PCR, όπως η ενίσχυση εκκινητών27,92 και αναντιστοιχίες αστάρι86. Ο προσεκτικός σχεδιασμός των εκκινητών μπορεί να βοηθήσει στην άμβλυνση αυτών των προβλημάτων, αλλά οι νέες παραλλαγές παραμένουν προβληματικές».
"Εμπλουτισμός στόχου. Οι μέθοδοι εμπλουτισμού στόχου (γνωστές και ως μέθοδοι pull-down, σύλληψης ή ειδικού εμπλουτισμού) μπορούν να χρησιμοποιηθούν για την αλληλουχία ολόκληρων ιικών γονιδιωμάτων απευθείας από κλινικά δείγματα χωρίς την ανάγκη προηγούμενης καλλιέργειας ή PCR98,99,100. Αυτές οι μέθοδοι συνήθως περιλαμβάνουν μικρούς ανιχνευτές RNA ή DNA που είναι συμπληρωματικοί προς την αλληλουχία αναφοράς παθογόνου (ή μια ομάδα αλληλουχιών αναφοράς).
«Η έλλειψη ενός βήματος καλλιέργειας σημαίνει ότι οι αλληλουχίες που λαμβάνονται είναι πιο αντιπροσωπευτικές του αρχικού ιού παρά των καλλιεργημένων απομονωμένων ιών και υπάρχουν λιγότερες μεταλλάξεις από ό, τι στα ενισχυμένα με PCR πρότυπα69,100. Η επιτυχία αυτής της μεθόδου εξαρτάται από τις διαθέσιμες αλληλουχίες αναφοράς για τον ιό που μας ενδιαφέρει· η ειδικότητα αυξάνεται όταν οι ανιχνευτές σχεδιάζονται έναντι μιας μεγαλύτερης ομάδας αλληλουχιών αναφοράς, καθώς αυτό οδηγεί σε καλύτερη σύλληψη της ποικιλομορφίας εντός και μεταξύ των δειγμάτων. Ο εμπλουτισμός του στόχου είναι δυνατός παρά τις μικρές αναντιστοιχίες μεταξύ προτύπου και ανιχνευτή. Ωστόσο, ενώ η ενίσχυση PCR απαιτεί μόνο γνώση των πλευρικών περιοχών μιας περιοχής-στόχου, ο εμπλουτισμός στόχου απαιτεί γνώση της εσωτερικής ακολουθίας για τον σχεδιασμό ανιχνευτών. Ωστόσο, εάν ένας ανιχνευτής αποτύχει, οι εσωτερικές και επικαλυπτόμενες περιοχές ενδέχεται να εξακολουθούν να καταγράφονται από άλλους ανιχνευτές69,100. Ο εμπλουτισμός στόχου δεν είναι κατάλληλος για τον χαρακτηρισμό νέων ιών που έχουν χαμηλή ομολογία σε γνωστούς ιούς για τους οποίους η μεταγονιδιωματική και, σε ορισμένες περιπτώσεις, η PCR χρησιμοποιώντας εκφυλισμένους εκκινητές, οι οποίοι είναι ένα μείγμα παρόμοιων αλλά μεταβλητών εκκινητών, μπορεί να είναι πιο κατάλληλοι.
«Τα αποτελέσματα αυτής της μελέτης, στην οποία αξιολογήθηκαν τρία διαφορετικά πρωτόκολλα εμπλουτισμού, δύο μεταγονιδιωματικές μέθοδοι και μία επικαλυπτόμενη μέθοδος PCR, έδειξαν ότι οι μεταγονιδιωματικές μέθοδοι ήταν οι λιγότερο ευαίσθητες, απέδωσαν τη χαμηλότερη κάλυψη γονιδιώματος για συγκρίσιμη προσπάθεια αλληλούχισης και ήταν πιο επιρρεπείς στο να οδηγήσουν σε ατελείς συναρμολογήσεις γονιδιώματος. Η μέθοδος PCR απαιτούσε επαναλαμβανόμενη ενίσχυση και ήταν πιο πιθανό να παραλείψει μικτές λοιμώξεις, αλλά όταν οι αντιδράσεις ήταν επιτυχείς είχε ως αποτέλεσμα το πιο σταθερό βάθος ανάγνωσης, ενώ το βάθος ανάγνωσης ήταν ανάλογο με τον αριθμό αντιγράφων του ιού στη μεταγονιδιωματική και τον εμπλουτισμό στόχου. Η PCR παρήγαγε περισσότερες ατελείς αλληλουχίες για ορισμένους γονότυπους HCV (ιδιαίτερα γονότυπου 2) από τη μεταγονιδιωματική και τον εμπλουτισμό στόχου. Ο εμπλουτισμός στόχου ήταν η πιο συνεπής μέθοδος για να οδηγήσει σε πλήρη γονιδιώματα και πανομοιότυπες αλληλουχίες συναίνεσης».
«Τα ζητήματα της ευαισθησίας και της μόλυνσης είναι ιδιαίτερα σημαντικά στο WGS, λόγω του κινδύνου τόσο ψευδώς αρνητικής όσο και ψευδώς θετικής ανίχνευσης παθογόνων παραγόντων. Η εξαιρετικά ευαίσθητη αλληλούχιση (είτε μεταγονιδιωματική, βασισμένη σε PCR ή με βάση τον εμπλουτισμό στόχου) μπορεί να ανιχνεύσει ιικά νουκλεϊκά οξέα χαμηλού επιπέδου μόλυνσης112,113. Για παράδειγμα, ιός λευχαιμίας ποντικού114,115 και αλληλουχίες παρόμοιες με παρβοϊούς116,117 είναι μόνο δύο από τους πολλούς ρύπους που μπορούν να προέλθουν από κοινά εργαστηριακά αντιδραστήρια, όπως στήλες εκχύλισης νουκλεϊκού οξέος118. Όπως και με άλλες εξαιρετικά ευαίσθητες τεχνολογίες, απαιτούνται ισχυρές εργαστηριακές πρακτικές και πρωτόκολλα για την ελαχιστοποίηση της μόλυνσης. Είναι επίσης σημαντικό να θυμόμαστε ότι η ανίχνευση του ιικού νουκλεϊκού οξέος δεν προσδιορίζει απαραίτητα την αιτία της ασθένειας και είναι καλή πρακτική όταν χρησιμοποιούνται μέθοδοι NGS για τη διάγνωση ιογενών λοιμώξεων να επιβεβαιώνονται τα ευρήματα με εναλλακτικές ανεξάρτητες μεθόδους που δεν βασίζονται σε δοκιμές για νουκλεϊνικά οξέα. Για παράδειγμα, σε περιπτώσεις εγκεφαλίτιδας άγνωστης αιτιολογίας, θετικά ευρήματα NGS μπορούν να επιβεβαιωθούν μέσω ανοσοϊστοχημικής ανάλυσης του προσβεβλημένου ιστού65,119, ή ταυτοποίηση του ιού με ηλεκτρονική μικροσκοπία ή ιστοκαλλιέργεια82.”
«Επί του παρόντος, η επιλογή της μεθόδου είναι συγκεκριμένη τόσο για τον ιό όσο και για το κλινικό ερώτημα. Η μεταγονιδιωματική αλληλούχιση είναι η πλέον κατάλληλη για διαγνωστική αλληλούχιση άγνωστων ή ελάχιστα χαρακτηρισμένων ιών, η αλληλούχιση αμπλικονίου PCR λειτουργεί καλά για βραχέα ιικά γονιδιώματα και χαμηλή ποικιλομορφία στις θέσεις δέσμευσης εκκινητών και ο εμπλουτισμός στόχου λειτουργεί για όλα τα μεγέθη παθογόνων, αλλά είναι ιδιαίτερα επωφελής για μεγάλους ιούς και για ιούς που έχουν ποικίλα αλλά καλά χαρακτηρισμένα γονιδιώματα. Υπάρχουν σήμερα δύο προφανείς τομείς καινοτομίας: μέθοδοι που μπορούν να καταστρέψουν αποτελεσματικά το DNA του ξενιστή χωρίς να επηρεαστεί το ιϊκό DNA και η περαιτέρω ανάπτυξη τεχνολογιών μακράς ανάγνωσης για την επίτευξη ευελιξίας και ανταγωνιστικής τιμολόγησης των τεχνολογιών σύντομης ανάγνωσης.
https://www.nature.com/articles/nrmicro.2016.182?WT.feed_name=subjects_bacteria
Αυτή η τρίτη πηγή είναι από τον Μάϊο του 2019. Επισημαίνει περαιτέρω ζητήματα όταν βασιζόμαστε στη γονιδιωματική για «ιούς». Επαναλαμβάνει το πρόβλημα της ύπαρξης χαμηλού «ιικού» υλικού σε σύγκριση με το DNA του ξενιστή καθώς και τη συνύπαρξη πολλών «παραλλαγών» μέσα στο ίδιο δείγμα. Επισημαίνει επίσης ορισμένα από τα ζητήματα της χρήσης PCR για την αλληλούχιση γονιδιωμάτων καθώς και την κρισιμότητα της επιλογής του σωστού γονιδιώματος αναφοράς:
Ένα πλήρες πρωτόκολλο για την αλληλούχιση ολόκληρου του γονιδιώματος του ιού από κλινικά δείγματα: Εφαρμογή στον κορωνοϊό OC43
"Η λήψη αλληλουχίας γονιδιώματος ιού απευθείας από κλινικά δείγματα εξακολουθεί να είναι ένα δύσκολο έργο λόγω του χαμηλού φορτίου γενετικού υλικού του ιού σε σύγκριση με το DNA του ξενιστή και της δυσκολίας να επιτευχθεί ακριβής συναρμολόγηση γονιδιώματος".
«Ωστόσο, παρά το σχετικά μικρό μέγεθος των γονιδιωμάτων του ιού, η αλληλούχισή τους συχνά παραμένει δύσκολη. Η μικρή ποσότητα γενετικού υλικού του ιού σε σύγκριση με το νουκλεϊκό οξύ του ξενιστή μειώνει την παραγωγή αλληλούχισης του ιού. Επιπλέον, πρέπει κανείς να αντιμετωπίσει τη δυσκολία ότι πολλές ιογενείς παραλλαγές συνυπάρχουν σε ένα μόνο δείγμα, παρουσιάζοντας περισσότερο ή λιγότερο μεταβλητές αλληλουχίες ανάλογα με τον εγγενή ρυθμό μετάλλαξης του ιού. Όλα αυτά τα σημεία επιβαρύνουν την αλληλούχιση και τη συναρμολόγηση του ιικού γονιδιώματος».
«Τρεις κύριες μέθοδοι που βασίζονται στο HTS χρησιμοποιούνται επί του παρόντος για την αλληλούχιση ολόκληρου του γονιδιώματος του ιού: μεταγονιδιωματική αλληλούχιση, αλληλούχιση εμπλουτισμού στόχου και αλληλούχιση PCR amplicon, καθεμία από τις οποίες παρουσιάζει οφέλη και μειονεκτήματα (Houldcroft et al., 2017). Στη μεταγονιδιωματική αλληλούχιση, το συνολικό DNA (ή/και RNA) από ένα δείγμα που περιλαμβάνει ξενιστές αλλά και βακτήρια, ιούς και μύκητεςεξάγεται και αλληλουχείται. Είναι μια απλή και οικονομικά αποδοτική προσέγγιση και είναι η μόνη προσέγγιση που δεν απαιτεί ακολουθίες αναφοράς. Αντ 'αυτού, οι άλλες δύο προσεγγίσεις HTS, ο εμπλουτισμός στόχου και η αλληλούχιση amplicon, εξαρτώνται και οι δύο από πληροφορίες αναφοράς για δολώματα σχεδιασμού ή εκκινητές. Ο περιορισμός της μεταγονιδιωματικής αλληλουχίας είναι ότι απαιτεί πολύ υψηλό βάθος αλληλούχισης για να ληφθεί αρκετό υλικό ιικού γονιδιώματος. Η αλληλούχιση εμπλουτισμού στόχου χρησιμοποιεί ολιγονουκλεοτίδια σύλληψης ειδικά για τον ιό για τον εμπλουτισμό της προετοιμασίας του ιικού γονιδιώματος πριν από την αλληλούχιση. Αυτή η μέθοδος είναι πιο συγκεκριμένη από τη μεταγονιδιωματική αλληλούχιση, αλλά συνεπάγεται υψηλότερο κόστος και πιο προηγμένη τεχνική εμπειρογνωμοσύνη για την προετοιμασία του δείγματος. Τέλος, η αλληλούχιση PCR amplicon είναι μια καθιερωμένη μέθοδος που συνίσταται σε ενίσχυση ειδικού ιικού γονιδιώματος με PCR πριν από την αλληλούχιση. Είναι εύκολα εφαρμόσιμο σε μεγάλο αριθμό δειγμάτων σε συνήθη χρήση και έτσι είναι πολύ κατάλληλο για κλινικά δείγματα. Η μέθοδος ενίσχυσης PCR, σε σύγκριση με τις άλλες, είναι ιδιαίτερα σημαντική για δείγματα που περιέχουν πολύ χαμηλό ιικό γενετικό υλικό, παρουσιάζει όμως αρκετά μειονεκτήματα. Η αλληλουχία του ιού που μας ενδιαφέρει πρέπει να είναι γνωστή και όχι πολύ μεταβλητή για να ενισχυθεί σωστά από το σύνολο των σχεδιασμένων εκκινητών. Μια δεύτερη παγίδα οφείλεται στο γεγονός ότι οι κύκλοι PCR μπορούν να εισαγάγουν ορισμένα σφάλματα ενίσχυσης κατά μήκος της ακολουθίας που καθιστούν το βήμα συναρμολόγησης πιο επιρρεπές σε λάθη. Τέλος, αυτή η μέθοδος μπορεί να χρησιμοποιηθεί μόνο για μικρά γονιδιώματα λόγω του αριθμού των αντιδράσεων PCR που πρέπει να περιοριστεί».
"Η ανάλυση βιοπληροφορικής των δεδομένων αλληλούχισης ιών βασίζεται συχνά στην ευθυγράμμιση ή τη χαρτογράφηση των αναγνώσεων έναντι μιας ακολουθίας αναφοράς που ακολουθείται από την εξαγωγή συναίνεσης με πλειοψηφική ψηφοφορία. Ωστόσο, το βήμα ευθυγράμμισης είναι γνωστό ότι εισάγει κάποιες προκαταλήψεις (Archer et al., 2010, Posada-Cespedes et al., 2017). Για παράδειγμα, εάν η μελετώμενη αλληλουχία ιών αποκλίνει από την επιλεγμένη αλληλουχία αναφοράς, οι αναγνώσεις που καλύπτουν τις περιοχές απόκλισης δεν θα μπορούσαν να ευθυγραμμιστούν σωστά, γεγονός που θα μεροληπτήσει την προκύπτουσα συναίνεση. Επιπλέον, το στάδιο χαρτογράφησης των αναγνώσεων σε αποκλίνουσες, επαναλαμβανόμενες ή χαμηλής πολυπλοκότητας περιοχές είναι ένα δύσκολο έργο που πρέπει να εξεταστεί προσεκτικά (Caboche et al., 2014). Τέλος, η επιλογή της ίδιας της ακολουθίας αναφοράς είναι ένα κρίσιμο βήμα από το οποίο θα εξαρτηθεί σε μεγάλο βαθμό η προκύπτουσα ακολουθία συναίνεσης.
https://www.sciencedirect.com/science/article/pii/S0042682219300728
Συνοπτικά (2016):

Οι αναλυτικές προσεγγίσεις για την ανακατασκευή και ταξινόμηση «ιϊκών» γονιδιωμάτων από μικτά δείγματα παραμένουν περιορισμένες ως προς την απόδοση και τη χρηστικότητα τους
Αξιοσημείωτες τεχνικές προκλήσεις εμπόδισαν την πρόοδο όπως:
Τα θραύσματα των «ιϊκών» γονιδιωμάτων είναι συνήθως τάξεις μεγέθους λιγότερο άφθονα από εκείνα του ξενιστή, των βακτηρίων ή / και άλλων οργανισμών σε κλινικά και περιβαλλοντικά μεταγονιδιώματα
Τα παρατηρούμενα «ιογενή» γονιδιώματα συχνά αποκλίνουν σημαντικά από τα γονιδιώματα αναφοράς, απαιτώντας τη χρήση εξαντλητικών προσεγγίσεων ευθυγράμμισης
Η υψηλή ενδοπληθυσμιακή «ιογενής» ποικιλομορφία μπορεί να οδηγήσει σε διφορούμενη ανακατασκευή αλληλουχίας
Τα σχετικά λίγα τεκμηριωμένα «ιικά» γονιδιώματα αναφοράς σε σύγκριση με τον εκτιμώμενο αριθμό διακριτών «ιικών» ταξινομικών κατηγοριών καθιστούν προβληματική την ταξινόμηση
Διάφορα εργαλεία λογισμικού έχουν αναπτυχθεί για να φιλοξενήσουν τις μοναδικές προκλήσεις και τις περιπτώσεις χρήσης που σχετίζονται με τον χαρακτηρισμό "ιογενών" ακολουθιών. Ωστόσο, η ποιότητα αυτών των εργαλείων ποικίλλει και η χρήση τους συχνά απαιτεί υπολογιστική εμπειρογνωμοσύνη ή πρόσβαση σε ισχυρούς υπολογιστές, περιορίζοντας έτσι τη χρησιμότητά τους σε πολλούς ερευνητές
Προς το παρόν έχουμε περιορισμένη αντίληψη της έκτασης της «ιογενούς» ποικιλότητας που υπάρχει στο περιβάλλον
Ένας ταξινομητής γρήγορης αλληλουχίας μπορεί να αποτύχει εντελώς να ανιχνεύσει ένα νέο στέλεχος ενός καλά χαρακτηρισμένου «ιού» και εξίσου μπορεί να αποδώσει καλά με τις ακολουθίες Illumina, αλλά να προσφέρει φτωχά αποτελέσματα για δεδομένα που παράγονται με την πλατφόρμα Ion Torrent
Τα περισσότερα λογισμικά ανάλυσης γονιδιωματικής αλληλουχίας δεν είναι κατάλληλα για «ιογενή» γονιδιώματα
Τα γενικά εργαλεία που είναι σε θέση να αντιμετωπίσουν τις προκλήσεις που θέτουν οι «ιογενείς» ακολουθίες είναι συχνά εφαρμόσιμα μόνο σε περιορισμένες περιπτώσεις
Η επιλογή μεταξύ προσεγγίσεων καθίσταται δύσκολη λόγω της αφθονίας ανόμοιων αλλά λειτουργικά ισοδύναμων μεθοδολογιών και γενικά της έλλειψης αυστηρών σημείων αναφοράς για «ιογενή» σύνολα δεδομένων
Η ευαίσθητη ανίχνευση προηγουμένως χαρακτηρισμένων «ιών» και «ιογενών» ανακαλύψεων παραμένουν βασικές προκλήσεις ανοιχτές για καινοτομία
Εντός των μεταγονιδιωμάτων η αναλογία των «ιικών» νουκλεϊνικών οξέων είναι συνήθως πολύ χαμηλότερη από εκείνη του ξενιστή ή άλλων μικροβίων, περιορίζοντας την ποσότητα του σήματος που είναι διαθέσιμο για ανάλυση μετά την αλληλούχιση
Η διήθηση μεγέθους ή ο εμπλουτισμός με βάση την πυκνότητα με φυγοκέντρηση είναι δύο αποτελεσματικές μέθοδοι για την αύξηση της απόδοσης "ιού", αν και τέτοιες μέθοδοι μπορεί να επηρεάσουν την παρατηρούμενη σύνθεση των "ιογενών" πληθυσμών
Η ενίσχυση PCR μπορεί να χρησιμοποιηθεί για τη δημιουργία αφθονίας συγκεκριμένων «ιογενών» αλληλουχιών που υπάρχουν σε ένα δείγμα, αν και ο αποτελεσματικός σχεδιασμός εκκινητών μπορεί να είναι δύσκολος παρουσία υψηλής γονιδιωματικής ποικιλότητας στα στοχευόμενα «ιικά» είδη
Η υπερβολική κάλυψη αλληλούχισης μπορεί να οδηγήσει στην κατασκευή υπερβολικά πολύπλοκων και δυσκίνητων de novo γραφημάτων συναρμολόγησης παρουσία υψηλής γονιδιωματικής ποικιλομορφίας, μειώνοντας την ποιότητα συναρμολόγησης
Χρησιμοποιώντας in silico (στον υπολογιστή) κανονικοποίηση, η υπερβολική κάλυψη μπορεί να μειωθεί με την απόρριψη αλληλουχιών που περιέχουν περιττές πληροφορίες
Μια άλλη in silico (στον υπολογιστή) στρατηγική για την αύξηση της αναλυτικής αποτελεσματικότητας με την απόρριψη περιττών δεδομένων είναι το φιλτράρισμα αλληλουχιών από γνωστούς άφθονους οργανισμούς μέσω ευθυγράμμισης με ένα ή περισσότερα γονιδιώματα αναφοράς χρησιμοποιώντας νάρθηκα ή ειδικό εργαλείο
Σε προσεγγίσεις που βασίζονται σε αναφορές, οι αναγνώσεις χαρτογραφούνται σε παρόμοιες περιοχές ενός παρεχόμενου γονιδιώματος προτύπου, μια καλά μελετημένη και υπολογιστικά αποτελεσματική διαδικασία που υλοποιείται με έναν δείκτη πίνακα επιθημάτων του γονιδιώματος αναφοράς
Αντίθετα, η de novo συναρμολόγηση είναι υπολογιστικά εξαντλητική αλλά σημαντική σε περιπτώσεις όπου είτε ένα γονιδίωμα-στόχος δεν χαρακτηρίζεται επαρκώς είτε επιδιώκεται η ανακατασκευή γονιδιωμάτων a priori άγνωστων οντοτήτων σε μεταγονιδιώματα, όπως σε μελέτες επιτήρησης
Η συναρμολόγηση είναι η διαδικασία λήψης μεγάλου αριθμού σύντομων αλληλουχιών DNA και η επανατοποθέτησή τους για να δημιουργηθεί μια αναπαράσταση των αρχικών χρωμοσωμάτων από τα οποία προήλθε το DNA (https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/genome-assembly)
Χρησιμοποιείται επίσης ευρέως για τη δημιουργία αλληλουχιών συναίνεσης ολόκληρου του γονιδιώματος για τη διευκόλυνση των αναλύσεων της «ιογενούς» παραλλαγής και είναι ένα τυπικό σημείο εκκίνησης για αναλύσεις διαφορετικών πληθυσμών καλά χαρακτηρισμένων «ιών»
Ακόμη και όταν υπάρχουν διαθέσιμες μεγάλες αναγνώσεις, η συναρμολόγηση παίζει σημαντικό ρόλο στον μετριασμό των υψηλών ποσοστών σφάλματος που σχετίζονται με τις τεχνολογίες αλληλούχισης ενός μορίου, αποδίδοντας ακριβείς αλληλουχίες συναίνεσης από ανακριβείς μεμονωμένες αναγνώσεις
Οι τυπικοί de novo συναρμολογητές έχουν σχεδιαστεί για να ανακατασκευάζουν γονιδιώματα με ομοιόμορφη κάλυψη αλληλούχισης σε όλο το μήκος τους, αλλά αυτό είναι προβληματικό για τα μεταγονιδιώματα (συμπεριλαμβανομένων των "viromes") όπου η κάλυψη συνήθως ποικίλλει σημαντικά τόσο μεταξύ διαφορετικών γονιδιωμάτων όσο και εντός μεμονωμένων γονιδιωμάτων
Η πολυπλοκότητα της ανακατασκευής πολλαπλών γονιδιωμάτων ορίζει ταυτόχρονα την τάση των συναρμολογητών μεταγονιδιώματος να καταρρέουν την παραλλαγή σε επίπεδο παραμόρφωσης ή κάτω από αυτό σε αλληλουχίες συναίνεσης
Η αποτελεσματικότητά τους μπορεί να είναι περιορισμένη ως συνέπεια της ακραίας διακύμανσης εντός συγκεκριμένων πληθυσμών «ιού» RNA λόγω μετάλλαξης και ανασυνδυασμού και της χαμηλής ή/και άνισης κάλυψης αλληλούχισης σε ένα συγκεκριμένο γονιδίωμα
Η de novo συναρμολόγηση είναι ιδιαίτερα ευαίσθητη στην ποιότητα των αλληλουχιών εισόδου, πράγμα που σημαίνει ότι τα προβλήματα κατά την εξαγωγή δειγμάτων, τον εμπλουτισμό και την προετοιμασία της βιβλιοθήκης μπορεί να είναι εξαιρετικά επιζήμια για τις κατάντη αναλύσεις
Τα «ιογενή» γονιδιώματα και τα μεταγονιδιώματα που περιλαμβάνουν υψηλή ενδοειδική παραλλαγή μπορεί να είναι δύσκολοι στόχοι για συναρμολόγηση, δημιουργώντας πολύπλοκα γραφήματα συναρμολόγησης και κατακερματισμένα συγκροτήματα
Ένας περιορισμός όλων των πιθανοτικών προσεγγίσεων ανασυγκρότησης πληθυσμών που βασίζονται στην ευθυγράμμιση, ωστόσο, είναι η εξάρτησή τους από μια ενιαία ακολουθία αναφοράς με την οποία θα εκτελεστεί η αρχική ευθυγράμμιση, μια διαδικασία που προϋποθέτει ένα βαθμό ομοιότητας αλληλουχίας που μπορεί να μην παρατηρείται πάντα σε διαφορετικές περιοχές
Όλες οι μέθοδοι ταξινόμησης, σε κάποιο βαθμό, εξαρτώνται από την ανίχνευση ομοιότητας μεταξύ μιας ακολουθίας ερωτημάτων και μιας συλλογής σχολιασμένων ακολουθιών
Οι υπολογιστικές απαιτήσεις των διαθέσιμων προσεγγίσεων ποικίλλουν δραματικά ανάλογα με την ικανότητά τους να ανιχνεύουν ομολογία σε αποκλίνουσες αλληλουχίες
Οι "ιογενείς" προσεγγίσεις αναγνώρισης εξαρτώνται συνήθως από αναζητήσεις ομοιότητας σε μια βάση δεδομένων χρησιμοποιώντας έναν νάρθηκα όπως το BLAST
Μπορούν να χρησιμοποιηθούν ολοκληρωμένες βάσεις δεδομένων (π.χ. GenBank) ή μικρότερες προσαρμοσμένες βάσεις δεδομένων που περιέχουν, για παράδειγμα, μόνο «ιογενείς» ακολουθίες ενδιαφέροντος, αν και οι τελευταίες μπορούν να παράγουν παραπλανητικά αποτελέσματα
Οι μέθοδοι τύπου BLAST περιορίζονται από:
Οι σχετικά λίγες επικυρωμένες «ιογενείς» αλληλουχίες κατατέθηκαν σε δημόσιες βάσεις δεδομένων
Η μεγάλη ποικιλομορφία μέσα στις «ιογενείς» οικογένειες που μπορεί να αποκρύψει τη συγγένεια
Η έλλειψη ενός καθορισμένου συνόλου βασικών γονιδίων κοινών σε όλους τους «ιούς» που μπορούν να χρησιμοποιηθούν για τη διάκριση των ειδών
Αυτά τα χαρακτηριστικά καθιστούν δύσκολη την εκχώρηση ορίων ομοιότητας για την ταξινόμηση που ισχύουν για όλους τους πιθανούς "ιούς" σε ένα δείγμα
Μια θεμελιώδης πρόκληση στην ταξινόμηση των «ιογενών» αλληλουχιών με οποιαδήποτε από αυτές τις μεθόδους παραμένει η περιορισμένη αναπαράστασή τους σε επιμελημένες βάσεις δεδομένων αλληλουχιών
Η τεκμηριωμένη κατανόησή μας για την έκταση της «ιογενούς» ποικιλομορφίας παραμένει επιφανειακή
Επομένως, οι αναγνώσεις της πραγματικής «ιογενούς» προέλευσης κινδυνεύουν να χαθούν σε πολλές περιπτώσεις
Η ταξινόμηση των «ιογενών» αλληλουχιών εξαρτάται σε μεγάλο βαθμό από την ποιότητα των επιμελημένων «ιογενών» βάσεων δεδομένων
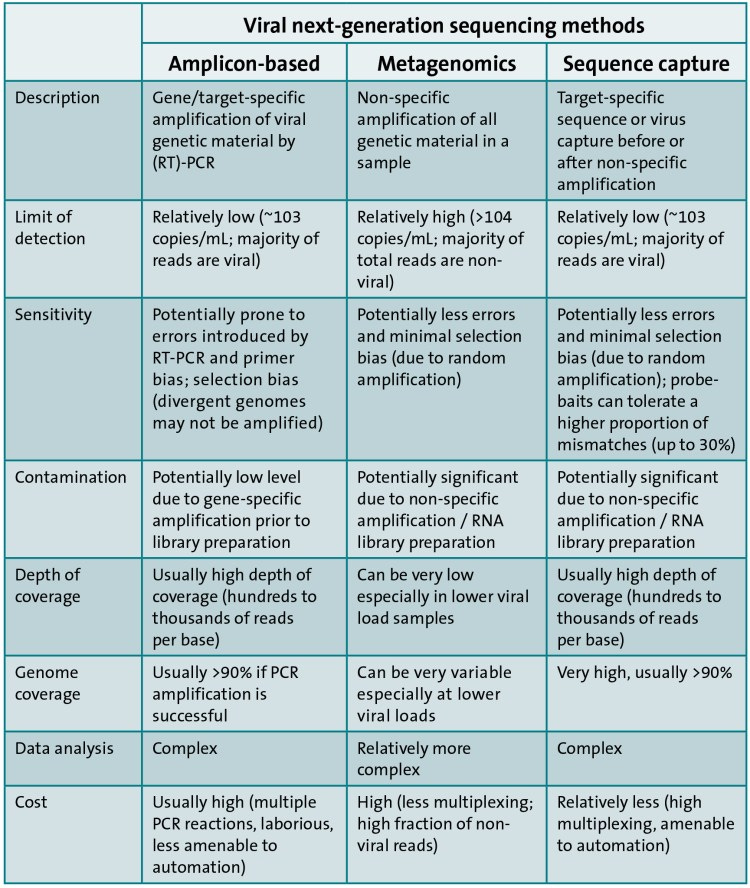
Συνοπτικά (2017):

Η αλληλούχιση των «ιικών» νουκλεϊνικών οξέων, είτε από καλλιέργειες είτε απευθείας από κλινικά δείγματα, περιπλέκεται από την παρουσία μολυσμένου DNA ξενιστή
Μεταγονιδιωματική:
Το συνολικό DNA ή / και το RNA, συμπεριλαμβανομένων των από τον ξενιστή, τα βακτήρια, τους «ιούς», τους μύκητες και άλλα παθογόνα, εξάγονται από ένα δείγμα και μια βιβλιοθήκη προετοιμάζεται και αλληλουχείται με αλληλούχιση κυνηγετικού όπλου ή αλληλούχιση RNA
Σε κλινικά δείγματα, η παρουσία μολυσματικών νουκλεϊνικών οξέων από τον ξενιστή και τους κοινούς μικροοργανισμούς μειώνει την ευαισθησία
Το ποσοστό των αναγνώσεων που ταιριάζουν με το γονιδίωμα του «ιού-στόχου» από το μεταγονιδιωματικό WGS είναι συχνά χαμηλό. για παράδειγμα, 0,008% για τον EBV στο αίμα ενός υγιούς ενήλικα, 0,0003% για τον «ιό» Lassa σε κλινικά δείγματα και 0,3% για τον «ιό» Zika σε δείγμα που εμπλουτίστηκε για σωματίδια «ιού» μέσω διήθησης και φυγοκέντρησης
Η συγκέντρωση σωματιδίων "ιού" από κλινικά δείγματα με έλξη με μεσολάβηση αντισωμάτων (για παράδειγμα, ανακάλυψη "ιού" βασισμένη σε συμπληρωματικό DNA (cDNA)-ενισχυμένο πολυμορφισμό μήκους θραυσμάτων (AFLP), συντομογραφία VIDISCA), διήθηση, υπερφυγοκέντρηση και εξάντληση των ελεύθερων νουκλεϊνικών οξέων, τα οποία προέρχονται κυρίως από τον ξενιστή, έχουν όλα δοκιμαστεί. Ωστόσο, αυτές οι μέθοδοι μπορούν επίσης να μειώσουν τη συνολική ποσότητα των «ιικών» νουκλεϊνικών οξέων, έτσι ώστε να είναι ανεπαρκής για την προετοιμασία μιας βιβλιοθήκης αλληλούχισης
Υπάρχουν μέθοδοι για την αύξηση της απόδοσης DNA, ωστόσο αυτές οι προσεγγίσεις είναι χρονοβόρες, δαπανηρές και μπορεί να αυξήσουν τον κίνδυνο προκαταλήψεων, σφαλμάτων και μόλυνσης, χωρίς απαραίτητα να βελτιώνουν την ευαισθησία και το ποσοστό των αναγνώσεων του ξενιστή συχνά παραμένει υψηλό
Είναι ζωτικής σημασίας να χρησιμοποιηθούν κατάλληλα εργαλεία βιοπληροφορικής και βάσεις δεδομένων που μπορούν να αξιολογήσουν εάν οι ανιχνευόμενες αλληλουχίες παθογόνων είναι πιθανό να είναι η αιτία μόλυνσης, τυχαίων ευρημάτων ή μολυσματικών παραγόντων
Τυχαία ευρήματα, τόσο σε ξενιστές όσο και σε μικροβιακές αλληλουχίες, μπορεί επίσης να παρουσιάσουν ηθικά και ακόμη και διαγνωστικά διλήμματα για την κλινική μεταγονιδιωματική
Εμπλουτισμός με αμπλικονικό PCR:
Η ενίσχυση PCR του «ιικού» γενετικού υλικού χρησιμοποιεί εκκινητές που είναι συμπληρωματικοί σε μια γνωστή νουκλεοτιδική αλληλουχία
Αλληλεπικαλυπτόμενα PCRs σε συνδυασμό με NGS έχουν χρησιμοποιηθεί για την αλληλουχία ολόκληρων γονιδιωμάτων μεγαλύτερων «ιών», αλλά αυτή η μέθοδος έχει περιορισμένη επεκτασιμότητα, καθώς απαιτούνται πολλοί εκκινητές και σχετικά μεγάλη ποσότητα αρχικού DNA που περιορίζει τον αριθμό των κατάλληλων δειγμάτων που είναι διαθέσιμα και επίσης τα γονιδιώματα που μπορούν να μελετηθούν χρησιμοποιώντας αυτή τη μέθοδο
Για κλινικές εφαρμογές αυτό είναι προβληματικό λόγω του υψηλού εργαστηριακού φόρτου εργασίας που σχετίζεται με πολυάριθμες διακριτές αντιδράσεις PCR, της ανάγκης για ατομική ομαλοποίηση των συγκεντρώσεων διαφορετικών ενισχυτικών PCR πριν από τη συγκέντρωση, της αυξανόμενης πιθανότητας αποτυχίας αντίδρασης λόγω αναντιστοιχίας εκκινητών (ιδιαίτερα για εξαιρετικά μεταβλητούς «ιούς») και του υψηλού κόστους εργασίας και αναλωσίμων
Ο αυξανόμενος αριθμός αντιδράσεων PCR απαιτεί αντίστοιχη αύξηση της ποσότητας του δείγματος και αυτό δεν είναι πάντα δυνατό καθώς τα κλινικά δείγματα είναι περιορισμένα
Τα εξαιρετικά μεταβλητά παθογόνα, ιδιαίτερα εκείνα που έχουν ευρέως αποκλίνουσες γενετικές γενεαλογίες ή γονότυπους, προκαλούν προβλήματα για την ενίσχυση PCR, όπως η ενίσχυση εκκινητών και οι αναντιστοιχίες εκκινητών
Ο προσεκτικός σχεδιασμός των εκκινητών μπορεί να βοηθήσει στην άμβλυνση αυτών των προβλημάτων, αλλά οι νέες παραλλαγές παραμένουν προβληματικές
Εμπλουτισμός-στόχος:
Μπορεί να χρησιμοποιηθεί για την αλληλουχία ολόκληρων «ιικών» γονιδιωμάτων απευθείας από κλινικά δείγματα χωρίς την ανάγκη προηγούμενης καλλιέργειας ή PCR, αλλά αυτές οι μέθοδοι συνήθως περιλαμβάνουν μικρούς ανιχνευτές RNA ή DNA που είναι συμπληρωματικοί προς την αλληλουχία αναφοράς παθογόνου (ή μια ομάδα αλληλουχιών αναφοράς)
Η επιτυχία αυτής της μεθόδου εξαρτάται από τις διαθέσιμες αλληλουχίες αναφοράς για τον "ιό" που μας ενδιαφέρει· η ειδικότητα αυξάνεται όταν οι ανιχνευτές σχεδιάζονται έναντι μιας μεγαλύτερης ομάδας αλληλουχιών αναφοράς, καθώς αυτό οδηγεί σε καλύτερη σύλληψη της ποικιλομορφίας εντός και μεταξύ των δειγμάτων
Ο εμπλουτισμός στόχου απαιτεί γνώση της εσωτερικής ακολουθίας για το σχεδιασμό ανιχνευτών
Ο εμπλουτισμός στόχου δεν είναι κατάλληλος για τον χαρακτηρισμό νέων «ιών» που έχουν χαμηλή ομολογία σε γνωστούς «ιούς»
Σε μία μελέτη, οι μεταγονιδιωματικές μέθοδοι ήταν οι λιγότερο ευαίσθητες, απέδωσαν τη χαμηλότερη κάλυψη γονιδιώματος για συγκρίσιμη προσπάθεια αλληλούχισης και ήταν πιο επιρρεπείς στο να οδηγήσουν σε ατελείς συναρμολογήσεις γονιδιώματος, ενώ η μέθοδος PCR απαιτούσε επαναλαμβανόμενη ενίσχυση και ήταν πιο πιθανό να χάσει μικτές λοιμώξεις
Τα ζητήματα της ευαισθησίας και της μόλυνσης είναι ιδιαίτερα σημαντικά στην WGS, λόγω του κινδύνου τόσο ψευδώς αρνητικής όσο και ψευδώς θετικής ανίχνευσης παθογόνων
Είναι σημαντικό να θυμόμαστε ότι η ανίχνευση του «ιικού» νουκλεϊκού οξέος δεν προσδιορίζει απαραίτητα την αιτία της ασθένειας και είναι καλή πρακτική όταν χρησιμοποιούνται μέθοδοι NGS για τη διάγνωση «ιογενών» λοιμώξεων να επιβεβαιώνονται τα ευρήματα με εναλλακτικές ανεξάρτητες μεθόδους που δεν βασίζονται στον έλεγχο για νουκλεϊκά οξέα
Απαιτούνται μέθοδοι που μπορούν να καταστρέψουν αποτελεσματικά το DNA του ξενιστή χωρίς να επηρεάσουν το «ιογενές» DNA
Συνοπτικά (2019):

Είναι αποδεκτό ότι η απόκτηση αλληλουχίας γονιδιώματος «ιού» απευθείας από κλινικά δείγματα εξακολουθεί να είναι ένα δύσκολο έργο λόγω του χαμηλού φορτίου γενετικού υλικού «ιού» σε σύγκριση με το DNA του ξενιστή και της δυσκολίας να επιτευχθεί ακριβής συναρμολόγηση γονιδιώματος
Παρά το σχετικά μικρό μέγεθος των γονιδιωμάτων του «ιού», η αλληλούχισή τους παραμένει συχνά δύσκολη
Η μικρή ποσότητα γενετικού υλικού "ιού" σε σύγκριση με το νουκλεϊκό οξύ του ξενιστή μειώνει την παραγωγή αλληλούχισης "viral"
Αρκετές «ιογενείς» παραλλαγές συνυπάρχουν σε ένα μόνο δείγμα, παρουσιάζοντας περισσότερο ή λιγότερο μεταβλητές αλληλουχίες
Στη μεταγονιδιωματική αλληλούχιση, εξάγεται και αλληλουχείται ολικό DNA (ή/και RNA) από ένα δείγμα που περιλαμβάνει ξενιστές αλλά και βακτήρια, «ιούς» και μύκητες
Ο εμπλουτισμός του στόχου και η αλληλούχιση του amplicon εξαρτώνται και οι δύο από τις πληροφορίες αναφοράς για τα δολώματα σχεδιασμού ή τους εκκινητές
Ο περιορισμός της μεταγονιδιωματικής αλληλουχίας είναι ότι απαιτεί πολύ υψηλό βάθος αλληλούχισης για να ληφθεί αρκετό «ιογενές» υλικό γονιδιώματος
Η PCR για αλληλούχιση παρουσιάζει αρκετά μειονεκτήματα όπως:
Η αλληλουχία του "ιού" ενδιαφέροντος πρέπει να είναι γνωστή και όχι πολύ μεταβλητή για να ενισχυθεί σωστά από το σύνολο των σχεδιασμένων εκκινητών
Μια δεύτερη παγίδα οφείλεται στο γεγονός ότι οι κύκλοι PCR μπορούν να εισαγάγουν ορισμένα σφάλματα ενίσχυσης κατά μήκος της ακολουθίας που καθιστούν το βήμα συναρμολόγησης πιο επιρρεπές σε λάθη
Τέλος, αυτή η μέθοδος μπορεί να χρησιμοποιηθεί μόνο για μικρά γονιδιώματα λόγω του αριθμού των αντιδράσεων PCR που πρέπει να περιοριστεί
Η βιοπληροφορική ανάλυση των δεδομένων αλληλούχισης "ιών" βασίζεται συχνά στην ευθυγράμμιση ή τη χαρτογράφηση των αναγνώσεων έναντι μιας ακολουθίας αναφοράς που ακολουθείται από την εξαγωγή συναίνεσης με πλειοψηφική ψηφοφορία
Το βήμα ευθυγράμμισης είναι γνωστό ότι εισάγει ορισμένες προκαταλήψεις
Εάν η μελετώμενη αλληλουχία "ιού" αποκλίνει από την επιλεγμένη ακολουθία αναφοράς, οι αναγνώσεις που καλύπτουν τις περιοχές απόκλισης δεν θα μπορούσαν να ευθυγραμμιστούν σωστά, γεγονός που θα μεροληπτήσει την προκύπτουσα συναίνεση
Η επιλογή της ίδιας της ακολουθίας αναφοράς είναι ένα κρίσιμο βήμα από το οποίο θα εξαρτηθεί σε μεγάλο βαθμό η προκύπτουσα ακολουθία συναίνεσης
Περιττό να πούμε ότι, προκειμένου η γονιδιωματική να είναι ένα πολύτιμο εργαλείο για τη μελέτη των «ιών», αυτοί οι «ιοί» πρέπει πρώτα να αποδειχθεί ότι υπάρχουν. Υπάρχουν πάρα πολλά λάθη, προκαταλήψεις, μεταβλητές και υπερβολική εξάρτηση από αναφορές και συναίνεση που καθιστούν ακατάλληλο να αποδειχθεί ή / και να μελετηθεί κάτι που δεν έχει καθαριστεί / απομονωθεί πρώτα.
Αυτό προειδοποίησε ο Charles Calisher. Αυτά τα νέα εργαλεία είναι αρκετά δροσερά και διασκεδαστικά, αλλά οι πληροφορίες που προέρχονται από αυτά δεν σημαίνουν τίποτα αν αγνοηθούν οι δοκιμασμένες και αληθινές μέθοδοι του παρελθόντος. Η σύγχρονη ιολογία έχει μόνο σειρές A, C, T, G σε μια βάση δεδομένων χωρίς τίποτα φυσικό να τις υποστηρίζει.
Δυστυχώς για τον Dr. Calisher, ακόμη και οι παλιές μέθοδοι ιολογίας που υπερασπίστηκε ήταν επιρρεπείς σε λάθη και δεν ήταν σε θέση να παράγουν την απόδειξη καθαρισμένων / απομονωμένων παθογόνων «ιών».
Αν σας άρεσε αυτό το άρθρο και θα θέλατε να βοηθήσετε να στηρίξετε το συνεχές έργο μου, ο παρακάτω σύνδεσμος είναι μια επιλογή.
Παρακαλώ βοηθήστε να στηρίξετε το έργο μου. 🙏

---Δικτυογραφία:
“Viral” Genomics: Nothing but Strings of Letters in a Data Bank – ViroLIEgy
https://viroliegy.com/2021/10/11/viral-genomics-nothing-but-strings-of-letters-in-a-data-bank/