
Τα Θραύσματα της Φαντασίας του SARS-CoV-2 - Πώς η Μαθηματική Απάτη και η Γενετική Αλληλουχία Κατασκεύασαν μια Παγκόσμια Κρίση

Σας ευχαριστώ θερμά για το ενδιαφέρον σας και την αναδημοσίευση των άρθρων μου. Θα εκτιμούσα ιδιαίτερα αν, κατά την κοινοποίηση, σ̲υ̲μ̲π̲ε̲ρ̲ι̲λ̲α̲μ̲β̲ά̲ν̲α̲τ̲ε̲ ̲κ̲α̲ι̲ ̲τ̲ο̲ν̲ ̲σ̲ύ̲ν̲δ̲ε̲σ̲μ̲ο̲ ̲(̲l̲i̲n̲k̲)̲ ̲τ̲ο̲υ̲ ̲ά̲ρ̲θ̲ρ̲ο̲υ̲ ̲μ̲ο̲υ̲. Αυτό όχι μόνο αναγνωρίζει την πηγή, αλλά επιτρέπει και σε άλλους να ανακαλύψουν περισσότερο περιεχόμενο. Η υποστήριξή σας είναι πολύτιμη για τη συνέχιση της δουλειάς μου.
Απόδοση στα ελληνικά: Απολλόδωρος - Michael Wallach | 5 Ιουλίου 2025
Μπορείτε να κάνετε εφάπαξ ή επαναλαμβανόμενες δωρεές μέσω του Ko-Fi:

Όταν θυμόμαστε ότι αυτή η εργασία του Fan Wu αποτέλεσε, στην ουσία, το θεμέλιο της «επιστημονικής» θεμελίωσης της ισχυριζόμενης πανδημίας, είναι δύσκολο να πούμε αν πρέπει να γελάσουμε ή να κλάψουμε. Με βάση το συμπέρασμα αυτής της εργασίας σχεδιάστηκε η δοκιμή PCR και ο κόσμος δοκιμάστηκε για αυτόν τον «νέο ιό». Πάνω σε αυτό το έγγραφο κατασκευάστηκαν συνθετικές αλληλουχίες «ιού» από εργαστήρια για να δοκιμαστεί ο “ιός” ως προς τις ιδιότητές του και να μελετηθεί η «φύση» του.
Με βάση αυτή την υποτιθέμενη αλληλουχία των Fan Wu et al. οι ειδήμονες των μέσων ενημέρωσης και οι απολογητές των ψευδοεπιστημόνων ισχυρίστηκαν ότι η μαθηματική πολυπλοκότητα είναι πέρα από την ικανότητα οποιουδήποτε εκτός του τομέα τους να την κατανοήσει ή να την σχολιάσει, και έτσι προσπάθησαν να σιγάσουν κάθε κριτική.
Και ήταν πάνω στα συμπεράσματα αυτής της δημοσίευσης που λέγεται ότι σχεδιάστηκε το υποτιθέμενο «εμβόλιο» και ότι δισεκατομμύρια άνθρωποι πιέστηκαν να κάνουν ενέσεις στον εαυτό τους. Ήταν το λογικό και μαθηματικό τέχνασμα στην καρδιά της πανδημίας.
- Michael Wallach
Φανταστείτε μια παγκόσμια κρίση χτισμένη πάνω σε ένα σπίτι από τραπουλόχαρτα, όπου το θεμέλιο δεν είναι ένας ιός αλλά ένα μαθηματικό ταχυδακτυλουργικό τέχνασμα. Στο άρθρο που ακολουθεί ο Michael Wallach αποκαλύπτει μια συγκλονιστική κριτική της δημοσίευσης του Fan Wu, του ακρογωνιαίου λίθου της αφήγησης για την πανδημία COVID-19. Ο Wallach εξηγεί ότι «ένας μαθηματικός από το Αμβούργο», ο οποίος εμβάθυνε στα δεδομένα πίσω από την υποτιθέμενη ανακάλυψη του SARS-CoV-2, τα βρήκε να καταρρέουν κάτω από την εξέταση -όπως ένας ντετέκτιβ που συνθέτει μια σκηνή εγκλήματος από τυχαία σκόνη της γειτονιάς, για να δηλώσει έναν φάντασμα ένοχο. Η διαδικασία, όπως περιγράφει λεπτομερώς ο Wallach, περιελάμβανε την ανάμειξη πνευμονικού υγρού σε εκατομμύρια γενετικά θραύσματα, τη διοχέτευσή τους σε έναν υπολογιστή προγραμματισμένο να «βρίσκει» έναν ιό και την παραγωγή μιας αλληλουχίας που δεν μπορούσε να αναπαραχθεί. Έως και το 17% αυτού του «ιού» προερχόταν από εργαστηριακές διαδικασίες και όχι από τον ασθενή, εκθέτοντας μια κυκλική λογική όπου τα στοιχεία ήταν εν μέρει κατασκευασμένα. Η αποκάλυψη του Wallach, που ενισχύεται από φωνές όπως ο Dr. Stefan Lanka, αμφισβητεί τους βασικούς ισχυρισμούς της ιολογίας, υποδηλώνοντας ότι η πανδημία στηρίχθηκε σε μια ψηφιακή ψευδαίσθηση. Ωστόσο, σημειώνει το Breaking the Spell, η εν λόγω αλληλουχία δεν είναι μοναδική στο COVID - η ιολογία στηρίζεται εδώ και καιρό σε ανεπιβεβαίωτες υποθέσεις, «μπερδεύοντας» τη συσχέτιση με την αιτιώδη συνάφεια. Οι επικριτές υποστηρίζουν ότι αυτή η μέθοδος παρακάμπτει την απομόνωση πραγματικών ιϊκών σωματιδίων, ένα ελάττωμα που επαναλαμβάνεται στο Beyond Contagion. Παρόλα αυτά, οι υπερασπιστές της ιολογίας επιμένουν ότι η γονιδιωματική πολυπλοκότητα επικυρώνει τα ευρήματά τους, απορρίπτοντας τους σκεπτικιστές ως παρείσακτους που δεν μπορούν να κατανοήσουν τα μαθηματικά.
Αυτή η ανάλυση αποτελεί φάρο για την αποκάλυψη όχι μόνο του COVID αλλά και του ευρύτερου οικοδομήματος της ιολογίας. Είναι ένα κάλεσμα αφύπνισης, που δείχνει πώς ένας τομέας μπορεί να οικοδομήσει μια παγκόσμια αφήγηση σε σαθρό έδαφος, όπως ένας σεφ που ισχυρίζεται μια συνταγή από μια σούπα τυχαίων συστατικών. Το No Pandemic υποστηρίζει ότι αυτό δεν ήταν απλώς ένα λάθος, αλλά μέρος ενός ενορχηστρωμένου σχεδίου, με το Σχέδιο για την Τυραννία να περιγράφει σκόπιμα βήματα για την εκμετάλλευση αυτής της επιστήμης για τον έλεγχο. Εν τω μεταξύ, το Hospitals, Not Viruses (Νοσοκομεία και Όχι Οι Ιοί) μετατοπίζει την προσοχή στα νοσοκομειακά πρωτόκολλα, όπως οι αναπνευστήρες, ως πραγματικούς παράγοντες θνησιμότητας και όχι σε έναν ιό. Η εργασία του μαθηματικού, όπως τονίζει ο Wallach, δείχνει ότι η ακολουθία της δημοσίευσης του Fan Wu θα μπορούσε εξίσου εύκολα να «βρει» τον HIV ή τον Έμπολα στο ίδιο δείγμα, αποκαλύπτοντας μια διαδικασία τόσο ελαττωματική που θα μπορούσε να δημιουργήσει οποιοδήποτε παθογόνο. Για κάθε ισχυρισμό για έναν νέο ιό, υπάρχει και ένας αντίλογος: δεν υπάρχουν πειράματα ελέγχου, δεν υπάρχουν απομονωμένοι ιοί, μόνο η καλύτερη εικασία ενός υπολογιστή. Αυτή η εισαγωγή θέτει τις βάσεις για τη βαθιά κατάδυση του Wallach, προσκαλώντας τους αναγνώστες να αμφισβητήσουν μια αφήγηση που αναδιαμόρφωσε τον κόσμο, οπλισμένοι με έναν φακό για να δουν μέσα από την ομίχλη της ψευδοεπιστήμης.
Με τις ευχαριστίες μας προς τον Michael Wallach.
Mike Wallach - Η Ψευδαίσθηση των Ιών
Η αναλογία των Ψευδών Στοιχείων του Ντετέκτιβ
Φανταστείτε ότι ένας διάσημος ντετέκτιβ καλείται να εξιχνιάσει ένα μυστηριώδες έγκλημα. Αντί να εξετάσει τον πραγματικό τόπο του εγκλήματος, ο ντετέκτιβ αποφασίζει να συλλέξει όλη τη σκόνη, τα συντρίμμια, τις τρίχες και τα τυχαία σωματίδια από ολόκληρη τη γειτονιά και να τα ρίξει σε ένα γιγαντιαίο μπλέντερ.
Αφού τα ανακατέψει όλα μαζί, ο ντετέκτιβ τροφοδοτεί αυτό το μείγμα σε ένα εξελιγμένο πρόγραμμα υπολογιστή που έχει σχεδιαστεί για να "βρίσκει μοτίβα". Ο υπολογιστής, εντυπωσιακά, καταφέρνει να συναρμολογήσει κομμάτια που θεωρητικά θα μπορούσαν να σχηματίσουν το προφίλ DNA ενός εγκληματία - αλλά εδώ είναι η παγίδα: ο υπολογιστής προγραμματίστηκε να ψάχνει ειδικά για μοτίβα που ταιριάζουν με προηγούμενες "λυμένες" υποθέσεις και θα συνεχίσει να αναδιατάσσει τα τυχαία συντρίμμια μέχρι να βρει κάτι που ταιριάζει.
Ο ντετέκτιβ τότε ανακοινώνει: "Βρήκα τον εγκληματία! Αυτή η αλληλουχία DNA το αποδεικνύει!" Όταν όμως άλλοι ντετέκτιβ προσπαθούν να επαναλάβουν την ίδια διαδικασία με τα ίδια συντρίμμια της γειτονιάς, παίρνουν εντελώς διαφορετικά αποτελέσματα. Ακόμα χειρότερα, όταν τρέχουν το πρόγραμμα αναζητώντας άλλους "γνωστούς εγκληματίες", βρίσκουν και γι' αυτούς στοιχεία - συμπεριλαμβανομένων εγκληματιών που έχουν πεθάνει εδώ και δεκαετίες.
Το πιο παράλογο μέρος; Ο ντετέκτιβ δεν βρήκε ποτέ στην πραγματικότητα έναν πραγματικό εγκληματία, δεν απομόνωσε ποτέ πραγματικά αποδεικτικά στοιχεία, και ολόκληρη η "αλληλουχία DNA" ήταν απλώς η καλύτερη εικασία ενός υπολογιστή για την τακτοποίηση της τυχαίας σκόνης της γειτονιάς. Ωστόσο, όλος ο κόσμος κλειδώνει επειδή το πρόγραμμα του υπολογιστή του ντετέκτιβ είπε ότι κυκλοφορεί ελεύθερος ένας επικίνδυνος εγκληματίας.
Αυτό είναι ουσιαστικά αυτό που συνέβη με την "ανακάλυψη" των ιών - μόνο που αντί για τα συντρίμμια της γειτονιάς, οι επιστήμονες αναμειγνύουν τα σωματικά υγρά αρρώστων ανθρώπων και αφήνουν τους υπολογιστές να οργανώσουν τα γενετικά θραύσματα σε μοτίβα που ονομάζουν "ιούς".
Η Εξήγηση του Ασανσέρ σε Ένα Λεπτό
Να τι ανακάλυψε αυτός ο μαθηματικός όταν έλεγξε την εργασία για το σπίτι πίσω από την πανδημία COVID:
Οι επιστήμονες ισχυρίστηκαν ότι βρήκαν έναν νέο ιό παίρνοντας υγρό από τους πνεύμονες ενός άρρωστου ασθενούς, αναμειγνύοντάς το σε εκατομμύρια γενετικά θραύσματα και βάζοντας έναν υπολογιστή να προσπαθήσει να συναρμολογήσει αυτά τα θραύσματα σαν παζλ. Ο υπολογιστής είπε τότε «Βρήκα έναν ιό!».
Αλλά όταν ο μαθηματικός μας προσπάθησε να το επαναλάβει αυτό σε έναν άλλο υπολογιστή χρησιμοποιώντας τα ίδια ακριβώς δεδομένα, δεν μπόρεσε να έχει το ίδιο αποτέλεσμα. Είναι σαν δύο αριθμομηχανές να δίνουν διαφορετικές απαντήσεις στο ίδιο μαθηματικό πρόβλημα - αυτό θα έπρεπε να είναι αδύνατο.
Ακόμα χειρότερα, όταν κοίταξε πιο προσεκτικά τι έκανε στην πραγματικότητα ο υπολογιστής, διαπίστωσε ότι απλώς τακτοποιούσε τυχαία γενετικά θραύσματα και ισχυριζόταν ότι σχημάτιζαν έναν ιό. Ο υπολογιστής θα μπορούσε εξίσου εύκολα να «βρει» τον HIV, τον Έμπολα ή οποιονδήποτε άλλο ιό στο ίδιο δείγμα. Είναι σαν ένα πρόγραμμα που μπορεί να «βρει» οποιαδήποτε λέξη θέλετε στην αλφαβητική σούπα.
Η πιο καταδικαστική ανακάλυψη; Έως και το 17% της αλληλουχίας του «ιού» προερχόταν από την ίδια την εργαστηριακή διαδικασία - που σημαίνει ότι εν μέρει έβρισκαν στοιχεία που είχαν δημιουργήσει οι ίδιοι και όχι στοιχεία που βρίσκονταν αρχικά στον άρρωστο ασθενή.
Το συμπέρασμα του μαθηματικού: Δεν υπάρχει καμία μαθηματική απόδειξη ότι ανακαλύφθηκε ποτέ στην πραγματικότητα ένας νέος ιός. Ο υπολογιστής απλώς έπαιζε ένα περίτεχνο παιχνίδι σύνδεσης των σημείων με τυχαία γενετικά θραύσματα.
[Κουδουνίζει το ασανσέρ]
Τα θέματα της έρευνας που μπορείτε να αναζητήσετε:
Αναζητήστε τον « Dr. Stefan Lanka » και τις δεκαετίες δουλειάς του που αμφισβητούν τις μεθόδους απομόνωσης ιών
Ψάξτε για το «The Viral Delusion documentary» για βαθύτερες καταδύσεις σε αυτά τα μεθοδολογικά προβλήματα
Διερευνήστε την «απομόνωση των ιών» έναντι της «γενετικής αλληλουχίας» - δύο πολύ διαφορετικές διαδικασίες που συχνά συγχέονται στο δημόσιο διάλογο
Αποκάλυψη: Η Γονιδιωματική Ακολουθία του SARS-CoV-2 είναι μια απάτη
Η Εργασία που ο Stefan Lanka Ήλπιζε ότι θα Άλλαζε τον Κόσμο
Michael Wallach | 1 Ιουνίου 2024
Στα τέλη του 2020, με τον κόσμο πλέον εντελώς κλειδωμένο και την απειλή των αναγκαστικών ή σχεδόν αναγκαστικών ενέσεων να αυξάνεται καθημερινά, ο εξαιρετικός Dr. Stefan Lanka, πρώην ιολόγος, έστειλε με ηλεκτρονικό ταχυδρομείο μια σύντομη εργασία ενός μαθηματικού στο Αμβούργο με εκπληκτικές συνέπειες.
Το έργο πολλών δεκαετιών του Dr. Lanka και των συναδέλφων του που αποκάλυπταν τα θεμελιώδη προβλήματα της ιολογίας βρίσκονταν τώρα στον απόηχο και στηρίζονταν σε μια μικρή ομάδα γιατρών, επιστημόνων, δημοσιογράφων και στοχαστών με ταχέως αυξανόμενο τρόπο το 2020, και οι αποκαλύψεις του άρχισαν να φτάνουν στο κοινό με σημαντικό τρόπο.
Ωστόσο, μεταξύ των απολογητών της ιολογίας αυξανόταν ένα ρεφρέν ενάντια σε πολλούς από τους ισχυρισμούς του Lanka. Το ρεφρέν ήταν απλό - ότι ίσως είχε δίκιο για την ψευδοεπιστήμη της προηγούμενης περιόδου της ιολογίας - αλλά ότι η πρόσφατη ιολογία ήταν πολύ πιο προηγμένη και βασιζόταν στη μαθηματική πολυπλοκότητα της γονιδιωματικής - μια πολυπλοκότητα που οι επικριτές απλά δεν μπορούσαν να κατανοήσουν.
Ο Dr Lanka, αποφασισμένος να δείξει ότι αυτή η φάρσα ήταν ακριβώς αυτό που ήταν - ότι η αποκαλούμενη γενετική αλληλουχία του «ιού» SARS-CoV-2 ήταν στην καλύτερη περίπτωση μια ψευδαίσθηση και στη χειρότερη μια απάτη - είχε προσεγγίσει έναν διακεκριμένο μαθηματικό για να απομακρύνει τον καπνό από αυτή τη μαθηματική πολυπλοκότητα που έκρυβε την απάτη πίσω από τους ισχυρισμούς ότι είχε ποτέ βρεθεί ένας «ιός» SARS-CoV-2.
Η εργασία στάλθηκε σε μια χούφτα φίλους του Lanka, συμπεριλαμβανομένου και εμού, αλλά με το Substack να βρίσκεται ακόμη στα σπάργανα και τους περισσότερους γιατρούς και επιστήμονες που ασκούν κριτική στο Covid να μην έχουν ακόμη ούτε καν ιστοσελίδα, με κάποιο τρόπο η εργασία δεν δημοσιεύτηκε ποτέ στο διαδίκτυο.
Παρακάτω είναι μια αναδημοσίευση ολόκληρης της εργασίας, που δημοσιεύεται εδώ στα ελληνικά, πιστεύω, για πρώτη φορά. Ελπίζω ότι μπορεί να συγκεντρώσει κάποια προσοχή από καθηγητές μαθηματικών, γενετιστές και απλούς ανθρώπους.
Ο μαθηματικός στον οποίο απευθύνθηκε ο Lanka, αποκαλώντας τον εαυτό του μόνο «Ένας μαθηματικός από το Αμβούργο» για να αποφύγει αντίποινα κατά της καριέρας του, εξέτασε την κεντρική ακαδημαϊκή εργασία που συντάχθηκε από τον διαβόητο πλέον Dr. Fan Wu et al στο Wuhan της Κίνας και τυπώθηκε στο τεύχος Φεβρουαρίου 2020 του περιοδικού Nature: «A new coronavirus associated with human respiratory disease in China» (Ένας νέος κορονοϊός που σχετίζεται με την ανθρώπινη αναπνευστική νόσο στην Κίνα), η οποία ισχυριζόταν ότι είχε γενετικά αλληλουχήσει έναν «νέο ιό» που αργότερα ονομάστηκε SARS-CoV-2.
Ο μαθηματικός στο Αμβούργο κατέβασε το πλήρες σύνολο δεδομένων και το κατάλληλο λογισμικό που είχε χρησιμοποιήσει ο Fan Wu για να ισχυριστεί την ανακάλυψη του SARS-CoV-2 και στη συνέχεια επανέλαβε τις διαδικασίες του Wu. Επέστρεψε στον Dr. Lanka μια σαφή διάψευση της βασικής συλλογιστικής που χρησιμοποιήθηκε για να συναχθεί το συμπέρασμα ότι ένας νέος ιός είχε ποτέ ανακαλυφθεί.
Για να κατανοήσει κανείς αυτό το εκπληκτικό έργο, πρέπει να κατανοήσει τις βασικές αρχές για το πώς ο Fan Wu και οι συνάδελφοί του ισχυρίστηκαν ποτέ ότι είχαν εξαρχής αλληλουχήσει έναν ιό. Αυτό που έκαναν δεν είναι α-παραδοσιακό στον τομέα της ιολογίας, αλλά μόλις το κατανοήσει κανείς, προκαλεί τη φαντασία του πώς μια τέτοια ακολουθία βημάτων θα μπορούσε ποτέ να γίνει αποδεκτή ως η κεντρική βάση πάνω στην οποία μπορεί να υποστηριχθεί οτιδήποτε, πόσο μάλλον ένας επιστημονικός τομέας, πόσο μάλλον η τρομοκράτηση και το κλείσιμο ολόκληρου του κόσμου.
Ένα μικρό ιστορικό: Μέχρι τη δεκαετία του 1980, η ιολογία δεν είχε ακόμη βρει και απομονώσει ούτε έναν ιό (ακόμη δεν έχει απομονώσει) και είχε αλλάξει ελάχιστα από τους θεμελιώδεις ισχυρισμούς της στη δεκαετία του 1950 ότι η τοποθέτηση μύξας αναμεμειγμένης με αντιβιοτικά σε νεφρικά κύτταρα πιθήκων αποδείκνυε την ύπαρξη ενός ιού στη μύξα εάν τα νεφρικά κύτταρα αλλοιώνονταν - αγνοώντας τους πολλούς άλλους λόγους για τους οποίους θα μπορούσε να λάβει χώρα μια τέτοια αλλοίωση. Η δεύτερη, και ειλικρινά, η μόνη άλλη σημαντική διαδικασία που γινόταν στην ιολογία εκείνη την εποχή ήταν η λήψη φωτογραφιών των νεκρωμένων μυξών κάτω από έναν ηλεκτρονικό μικρογράφο. Εάν οι «ιολόγοι» έβλεπαν κύκλους (ή άλλο προκαθορισμένο σχήμα) στις εικόνες, ισχυρίζονταν ότι αυτό ήταν μια περαιτέρω απόδειξη ότι είχε βρεθεί ένας “ιός” - αγνοώντας και πάλι το πρόβλημα ότι δεν είχαν κανένα λόγο να συμπεράνουν ότι οι θεωρητικοί τους «ιοί» ήταν ο μόνος πιθανός λόγος για τον οποίο κάποιος θα μπορούσε να δει έναν κύκλο ή ένα άλλο προκαθορισμένο σχήμα.
Η προφανής αναποτελεσματικότητα και τα κενά λογικά σφάλματα αυτών των «πειραμάτων» είχαν ίσως αρχίσει να εξαντλούνται και ο τομέας είχε κάνει λίγες προόδους στη φαντασία της δημόσιας σφαίρας.
Όταν η επανάσταση των ηλεκτρονικών υπολογιστών εμφανίστηκε ταυτόχρονα με τη μελέτη της γονιδιωματικής, η ιολογία έψαχνε να βρει έναν τρόπο να μελετήσει τα θεωρητικά της (αλλά ακόμη δεν είχε βρεθεί, απομονωθεί ή αποδειχθεί η ύπαρξή τους) σωματίδια χρησιμοποιώντας τη νέα τεχνολογία.
Αξίζει να σημειωθεί ότι επρόκειτο για μια εντελώς διαφορετική διαδικασία από αυτή που χρησιμοποιήθηκε γενικότερα στη γονιδιωματική. Σε άλλα πεδία της γονιδιωματικής, ξεκινούσε κανείς με ένα πραγματικό απομονωμένο δείγμα του εν λόγω υλικού (π.χ. ένα ΑΛΟΓΟ, μια ΜΥΓΑ, ή ένα στέλεχος ΒΑΚΤΗΡΙΟΥ κ.λπ.) και κατέγραφε τι RNA θα μπορούσε να βρεθεί με συνέπεια στο απομονωμένο δείγμα αυτού του υλικού. Ωστόσο, στην ιολογία, δεδομένου ότι ποτέ δεν είχαν ένα πραγματικό δείγμα του εν λόγω «ιού», απομονωμένο από το υπόλοιπο ανθρώπινο υγρό, το μόνο που μπορούσαν να κάνουν ήταν να καταγράψουν το σύνολο του γενετικού υλικού στα δείγματα των μυξών τους, και στη συνέχεια να κάνουν εικασίες για το από τι θα μπορούσε να αποτελείται ο φανταστικός τους ιός.
Τα τελευταία σαράντα χρόνια, αυτή η λεγόμενη «γενετική αλληλουχία» έχει γίνει η κεντρική διαδικασία με την οποία οι άνθρωποι με τις εργαστηριακές ποδιές (δεν μπορώ να πείσω τον εαυτό μου να τους αποκαλέσω επιστήμονες) ισχυρίζονται από τότε ότι ανακάλυψαν νέους «ιούς». Ουσιαστικά, τα βήματα είναι τα εξής:
Βρείτε ένα άτομο που είναι άρρωστο, ΥΠΟΘΕΣΤΕ ότι είναι άρρωστο λόγω ενός ιού, και στη συνέχεια πάρτε ένα δείγμα από τις μύξες ή το «πνευμονικό υγρό» του.
Συνδυάστε αυτό το υγρό με αλατόνερο και αντιβιοτικά (και συχνά πολλά άλλα συστατικά).
Τροφοδοτήστε αυτό το υγρό μείγμα σε μια μηχανή η οποία διασπά το υλικό σε δεκάδες εκατομμύρια γενετικά θραύσματα.
Υποβάλετε αυτό το μείγμα σε μια διαδικασία PCR για την ενίσχυση του αριθμού των θραυσμάτων RNA, συμπεριλαμβανομένης της «ενίσχυσης» τυχόν συγκεκριμένων θραυσμάτων RNA που οι ιολόγοι αναμένουν να βρουν.
Βάλτε αυτό το μηχάνημα-υπολογιστή να δημιουργήσει έναν κατάλογο αυτών των γενετικών θραυσμάτων.
Βάλτε τον υπολογιστή να αποκλείσει έναν (καθόλου πειστικό) μερικό κατάλογο γνωστών ανθρώπινων ενδογενών θραυσμάτων από το σύνολο δεδομένων του.
Βάλτε τον υπολογιστή να χρησιμοποιήσει αλγορίθμους πιθανοτήτων για να βρει αλληλουχίες θραυσμάτων που επικαλύπτονται και δημιουργούν πιθανά «contigs» - στη συνέχεια, επιλέξτε τις μακρύτερες αλληλουχίες από αυτές που μπορούν θεωρητικά να ενωθούν από τον υπολογιστή.
Βάλτε τον υπολογιστή να εξάγει έναν κατάλογο αυτών των συνδυασμών που μοιάζουν περισσότερο με αλληλουχίες που επίσης δημιουργήθηκαν υποθετικά και αποδόθηκαν σε φανταστικούς «ιούς» στο παρελθόν".
Στη συνέχεια, οι ιολόγοι επιλέγουν μεταξύ αυτών των συνδυασμών, αποφασίζοντας με συναίνεση, τη μία θεωρητική ακολουθία του υπολογιστή που πιστεύουν ότι είναι ο ιός που αρρωσταίνει τον ασθενή. Εάν δεν μπορούν να προσεγγίσουν έστω και στο ελάχιστο την αντιστοίχιση μιας από τις αλληλουχίες που συνέθεσε ο υπολογιστής με μια προηγουμένως θεωρητική αλληλουχία, τότε οι ιολόγοι ισχυρίζονται ότι αυτό που βρήκαν πρέπει να είναι ένας «νέος ιός» και επιλέγουν με συναίνεση μεταξύ των καταγεγραμμένων συνδυασμών που εκδίδει ο υπολογιστής αυτόν που υποθέτουν καλύτερα ότι είναι ο ιός (και όχι απλώς μια ανούσια όξινη ορολογία).
Για όσους θέλουν μια βαθύτερη κατάδυση σε αυτή την ανοησία, το καλύπτω σε βάθος στο ντοκιμαντέρ The Viral Delusion στο www.theviraldelusion.com, ο Mike Stone το καλύπτει με μεγάλη λεπτομέρεια στο blog του viroliegy.com, ο Dr. Mark Bailey το ξεσκίζει στο άρθρο του «A Farewell To Virology» και ο Dr. Tom Cowan , Dr. Andy Kaufman και η Amandha Volmer (μεταξύ άλλων) έχουν αμέτρητες ώρες που περιγράφουν λεπτομερώς τον παραλογισμό αυτού του πράγματος στα βίντεο τους. Μπορείτε φυσικά να διαβάσετε και το αρχικό έγγραφο του Fan Wu για να δείτε το περίγραμμα αυτών των βημάτων. Όμως, όπως καθιστά σαφές το παρακάτω έγγραφο, ακόμη και οι πολλοί γιατροί και επιστήμονες με τους οποίους μίλησα στο The Viral Delusion υποτίμησαν τις μαθηματικές ανοησίες που χρησιμοποιήθηκαν στο έγγραφο του Fan Wu (αλλά θάφτηκαν βαθιά στην ενότητα της μεθοδολογίας) - ανοησίες τις οποίες ο αξιότιμος μαθηματικός μας αποκαλύπτει παρακάτω.
Φυσικά, μπορώ ήδη να σας ακούσω να φωνάζετε - σταματήστε, περιμένετε! Δεν χρειάζεται να προχωρήσουμε περαιτέρω. Πρόκειται ήδη για μια σειρά βημάτων που στερείται γελοιωδώς μεθοδολογικής ορθότητας. Ναι, το ξέρω. Για να γίνει το θέμα για τους νέους αναγνώστες πιο ξεκάθαρο, θα μπορούσε κανείς να χρησιμοποιήσει την ίδια σειρά βημάτων για να καταγγείλει την ανακάλυψη οποιασδήποτε νέας γενετικής ακολουθίας - είτε πρόκειται για «ιό» είτε για «σημάδι του διαβόλου», «ψείρες» ή απόδειξη της ανακάλυψης των γονιδίων ενός εξωγήινου. Πρόκειται για ένα χαρακτηριστικό παράδειγμα ψευδοεπιστήμης που βασίζεται στο λογικό τέχνασμα που είναι γνωστό ως «begging the question» (προκαλώ το ερώτημα λαμβάνοντας το ως δεδομένο). Και αυτό είναι μόνο η κορυφή του παγόβουνου των λογικών προβλημάτων με την εξαγωγή οποιουδήποτε είδους συμπεράσματος με βάση τα παραπάνω βήματα.
Αλλά ας συνεχίσουμε - γιατί το ρεφρέν που ακούγεται σταθερά από τους απολογητές της ιολογίας ήταν ότι κανένα από αυτά τα λογικά προβλήματα δεν είχε σημασία, τα μαθηματικά ήταν τόσο πολύπλοκα και τόσο βαθιά που αποδείκνυαν ότι η ιολογία είχε δίκιο από την αρχή, και όποιος το αμφισβητούσε απλώς δεν μπορούσε να καταλάβει.
Έρχεται ο μαθηματικός από το Αμβούργο. Η εργασία του βρίσκεται παρακάτω, και είστε ευπρόσδεκτοι να την προσπεράσετε φυσικά. Αλλά είναι γραμμένη σε μάλλον μεθυστική γλώσσα, οπότε θα αφιερώσω λίγο χρόνο για να την συνοψίσω εδώ.
Όπως θα δείτε, ο μαθηματικός ξεκίνησε την ανάλυσή του κατεβάζοντας το σύνολο των δεδομένων του πλήρους κατακερματισμού του RNA από το αρχικό πείραμα και προσπαθώντας να επαναλάβει απλώς τα βήματα του υπολογιστή που έγιναν στην εργασία.
Διαπίστωσε ότι ακόμη και αυτά τα βήματα δεν ήταν δυνατό να αναπαραχθούν από έναν υπολογιστή. Οι αλληλουχίες που εξάγονται από το λογισμικό το οποίο ισχυριζόταν ότι βρήκε τον «SARS-CoV-2» δεν μπορούσαν να εξαχθούν από άλλον υπολογιστή που εκτελούσε το ίδιο λογισμικό.
Αυτό δεν είναι μικρό θέμα! Όπως οι περισσότεροι γνωρίζουν, η βασική επιστημονική αυστηρότητα απαιτεί ότι τα πειράματα πρέπει να είναι αναπαραγώγιμα για να θεωρηθούν έγκυρα τα συμπεράσματά τους - αλλά η μη αναπαραγώγιμη φύση της αλληλουχίας SARS-CoV-2 υπερβαίνει κατά πολύ αυτό. Δεν μιλάμε για τη δυνατότητα αναπαραγωγής ενός πειράματος που συνέβη στη ζωντανή φύση- μιλάμε για έναν υπολογιστή που εκτελεί το ίδιο λογισμικό πάνω στο ίδιο σύνολο δεδομένων και δεν είναι σε θέση να αναπαράγει αυτό που ισχυρίστηκε ότι συνέβη σε έναν άλλο υπολογιστή που εκτελεί το ίδιο λογισμικό πάνω στο ίδιο σύνολο δεδομένων!
Για να γίνει αυτό σαφές, είναι σαν να ισχυρίζονταν οι Fan Wu et al ότι ο υπολογιστής τους μπορούσε να συλλαβίσει μια λέξη από ένα σετ Scrabble με περισσότερα «P» από όσα περιείχε το σετ Scrabble.
Ωστόσο, αυτό είναι μόνο η αρχή. Ο μαθηματικός προχώρησε και υπέθεσε ότι το αρχικό σύνολο δεδομένων και το αρχικό αποτέλεσμα ήταν σωστά για να συνεχίσει την ανάλυσή του.
Αυτό που βρήκε αποκαλύπτει ότι οποιοδήποτε συμπέρασμα βασισμένο σε αυτά τα δεδομένα ότι ο ασθενής που μελετήθηκε στο Wuhan είχε έναν νέο ιό ήταν εντελώς αβάσιμο.
Κατ' αρχάς, και πάλι, καθιστά σαφές ότι οι αλληλουχίες που ισχυρίστηκαν ότι έβγαλαν οι Fan Wu et al. ΔΕΝ μπορούσαν να συναρμολογηθούν από τα κομμάτια RNA που καταγράφηκαν από τον υπολογιστή στο δείγμα του ασθενούς.
Δεύτερον, διαπίστωσε ότι δεν υπήρχε κανένας τρόπος να πει αν η συναρμολογημένη αλληλουχία (που αργότερα ονομάστηκε SARS-CoV-2) προερχόταν από ανθρώπινο ή μη ανθρώπινο RNA. Με άλλα λόγια, δεν υπάρχει τίποτα στο πείραμα που να δείχνει αν η αλληλουχία συναρμολογήθηκε ΑΠΟ έναν «ιό» στο δείγμα ή απλώς από τυχαία κομμάτια RNA στο δείγμα.
Τρίτον, διαπίστωσε ότι δεν υπήρχε κανένας τρόπος να πει αν η συναρμολογημένη αλληλουχία προερχόταν από πραγματικά υπάρχον RNA στο δείγμα ή αν είχε καταρτιστεί από δείκτες RNA που υπήρχαν απλώς ως υποπροϊόν της ενίσχυσης με PCR στο δείγμα.
Τέταρτον, διαπίστωσε ότι έως και το 17% της τελικής αλληλουχίας βασιζόταν σε RNA contigs που είχαν στοχευθεί ειδικά και στη συνέχεια «βρέθηκαν» από τη διαδικασία PCR σε κατώφλια κύκλων ct 35 έως 45, αριθμοί κύκλων που είναι καλά γνωστοί στη βιβλιογραφία για να «βρίσκεις» ό,τι θέλεις.
Πέμπτον, διαπίστωσε ότι αυτά τα contigs ήταν σημαντικά πιο πιθανό να προέρχονται από την ίδια τη διαδικασία PCR παρά από το αρχικό δείγμα και ότι ήταν ΠΟΛΥ απίθανο να προέρχονται όλα τα contigs της αλληλουχίας SARS-CoV-2 (ή ακόμη και τα περισσότερα) από το αρχικό δείγμα.
Έκτον, διαπίστωσε ότι τα contigs στο εναπομείναν δείγμα δεδομένων ΑΦΟΥ η δημοσίευση Fan Wu ισχυρίζεται ότι το φιλτράρισε για γνωστό ανθρώπινο RNA, ταίριαζαν με γνωστό ανθρώπινο RNA.
Έβδομον, διαπίστωσε ότι η τελική αλληλουχία (SARS-CoV-2) που ισχυρίστηκε ότι ταιριάζει με «ιούς της κορώνας» δεν ταίριαζε καν με αυτές τις θεωρητικές αλληλουχίες, εκτός εάν συμπεριλήφθηκε ένα «ποσοστό σφάλματος» που ήταν πάνω από 10 τοις εκατό.
Όγδοο, επιδίωξε να ανακαλύψει αν θα μπορούσε κανείς να πάρει το δείγμα και να «βρει» άλλους ισχυριζόμενους ιούς σε αυτό. Έψαξε για «Ηπατίτιδα» και “HIV” και βρήκε ότι ΚΑΙ οι δύο είχαν χαμηλότερα ποσοστά σφάλματος από τον «SARS-CoV-2».
Ένατο, έψαξε για τις ισχυριζόμενες αλληλουχίες του «ιού Έμπολα» και του «ιού Μάρμπουργκ» και «βρήκε» και αυτές στο δείγμα, με συγκρίσιμα ποσοστά σφάλματος με τον «SARS-CoV-2».
Δέκατον, διαπίστωσε ότι δεν διεξήχθησαν πειράματα ελέγχου για να αποκλειστεί οποιοδήποτε από τα παραπάνω ή άλλα ενδεχόμενα.
Συμπερασματικά, ο μαθηματικός γράφει ότι: «μπορέσαμε να τεκμηριώσουμε την υπόθεσή μας ότι οι ισχυριζόμενες αλληλουχίες ιικού γονιδιώματος είναι παρερμηνείες με την έννοια ότι έχουν κατασκευαστεί ή κατασκευάζονται απαρατήρητες από μη ιϊκά τμήματα νουκλεϊκών οξέων».
Με άλλα λόγια, δεν υπάρχει τίποτα στα μαθηματικά που να υποδηλώνει το συμπέρασμα ότι είχε βρεθεί ένας νέος ιός ή ότι ήταν με οποιονδήποτε τρόπο η αιτία της ασθένειας του αρχικού άνδρα - στην πραγματικότητα συμβαίνει το αντίθετο - είναι ΠΙΟ πιθανό με βάση τα δεδομένα ότι η αλληλουχία που συνέταξε ο υπολογιστής και που ο Fan Wu ισχυρίστηκε ότι ήταν «SARS-CoV-2» δεν προερχόταν από «ιό».
Μια προσεκτική ανάγνωση του εγγράφου του μαθηματικού πράγματι υποδηλώνει και εξηγεί ότι είναι πολύ ΠΙΟ πιθανό η αλληλουχία «SARS-CoV-2» να δημιουργήθηκε από τυχαία κομμάτια RNA που επιπλέουν στο δείγμα σε συνδυασμό με ειδικά δημιουργημένες “ανακαλύψεις” τμημάτων RNA που δημιουργήθηκαν με PCR με σκοπό ακριβώς την «εύρεσή» τους.
Όταν θυμόμαστε ότι αυτή η εργασία του Fan Wu αποτέλεσε, στην ουσία, το θεμέλιο της «επιστημονικής» θεμελίωσης της ισχυριζόμενης πανδημίας, είναι δύσκολο να πούμε αν πρέπει να γελάσουμε ή να κλάψουμε. Με βάση το συμπέρασμα αυτής της εργασίας σχεδιάστηκε η δοκιμή PCR και ο κόσμος δοκιμάστηκε για αυτόν τον «νέο ιό». Πάνω σε αυτό το έγγραφο κατασκευάστηκαν συνθετικές αλληλουχίες «ιού» από εργαστήρια για να δοκιμαστεί ο “ιός” ως προς τις ιδιότητές του και να μελετηθεί η «φύση» του.
Με αφορμή αυτή την ισχυριζόμενη ακολουθία από τους Fan Wu κ.ά., οι ειδήμονες των μέσων ενημέρωσης και οι απολογητές των ψευδοεπιστημόνων ισχυρίστηκαν ότι η μαθηματική πολυπλοκότητα είναι πέρα από την ικανότητα οποιουδήποτε εκτός του τομέα τους να την κατανοήσει ή να την σχολιάσει, και έτσι προσπάθησαν να κλείσουν κάθε κριτική.
Και ήταν πάνω στα συμπεράσματα αυτής της δημοσίευσης που λέγεται ότι σχεδιάστηκε το υποτιθέμενο «εμβόλιο» και ότι δισεκατομμύρια άνθρωποι πιέστηκαν να κάνουν ενέσεις στον εαυτό τους. Ήταν το λογικό και μαθηματικό τέχνασμα στην καρδιά της πανδημίας.
Αλλά για να μην κλέψω περαιτέρω την αίγλη από την ανάλυσή του, παραθέτω παρακάτω την αναδημοσίευση της εργασίας του μαθηματικού.
Μοιραστείτε τις σκέψεις σας μετά την ανάγνωση (και ένα μεγάλο ευχαριστώ στην Petra Liverani για τη σωστή μορφοποίηση της εργασίας στο Substack!)
Δομική ανάλυση δεδομένων ακολουθιών στην ιολογία
Μια στοιχειώδης προσέγγιση με παράδειγμα τον SARS-CoV-2
Συγγραφέας
Από έναν μαθηματικό από το Αμβούργο, ο οποίος θα ήθελε να παραμείνει άγνωστος
Περίληψη
Η de novo μετα-τρανσκριπτονομική αλληλουχία ή η αλληλουχία ολόκληρου του γονιδιώματος είναι αποδεκτές μέθοδοι στην ιολογία για την ανίχνευση ισχυριζόμενων παθογόνων ιών. Κατά τη διαδικασία αυτή, δεν ανιχνεύονται σωματίδια ιών (ιούς) και με την έννοια της λέξης απομόνωση, απομονώνονται και χαρακτηρίζονται βιοχημικά. Στην περίπτωση του SARS-CoV-2, το ολικό RNA συχνά εξάγεται από δείγματα ασθενών (π.χ.: υγρό βρογχοκυψελιδικού καθαρισμού (BALF) ή επιχρίσματα λαιμού-ρινός) και αλληλουχίζεται. Ειδικότερα, δεν υπάρχουν ενδείξεις ότι τα τμήματα RNA που χρησιμοποιούνται για τον υπολογισμό των αλληλουχιών του ιικού γονιδιώματος είναι ιικής προέλευσης.
Ως εκ τούτου, εξετάσαμε τη δημοσίευση «A new coronavirus associated with human respiratory disease in China» [1] και τα σχετικά δημοσιευμένα δεδομένα αλληλουχίας με αναγνωριστικό βιοέργου PRJNA603194 με ημερομηνία 27/01/2020 για την αρχική πρόταση γονιδιακής αλληλουχίας του SARS-CoV-2 (GenBank: MN908947.3). Μια επανάληψη της de novo συναρμολόγησης με το Megahit (v.1.2.9) έδειξε ότι τα δημοσιευμένα αποτελέσματα δεν μπορούσαν να αναπαραχθούν. Ενδέχεται να ανιχνεύσαμε (ριβοσωμικά) ριβονουκλεϊκά οξέα ανθρώπινης προέλευσης, σε αντίθεση με ό,τι αναφέρθηκε στο [1]. Περαιτέρω ανάλυση παρείχε ενδείξεις για πιθανή μη ειδική ενίσχυση των αναγνώσεων κατά την επιβεβαίωση της PCR και τον προσδιορισμό γονιδιωματικών άκρων που δεν σχετίζονται με τον SARS-CoV-2 (MN908947.3).
Τέλος, πραγματοποιήσαμε ορισμένες συναρμολογήσεις βάσει αναφοράς με πρόσθετες αλληλουχίες γονιδιωμάτων όπως ο SARS-CoV, ο ιός της ανθρώπινης ανοσοανεπάρκειας, ο ιός της δέλτα ηπατίτιδας, ο ιός της ιλαράς, ο ιός Zika, ο ιός Ebola ή ο ιός Marburg για να μελετήσουμε τη δομική ομοιότητα των παρόντων δεδομένων αλληλουχιών με τις αντίστοιχες αλληλουχίες. Λάβαμε προκαταρκτικές ενδείξεις ότι ορισμένες από τις αλληλουχίες του ιικού γονιδιώματος που μελετήσαμε στην παρούσα εργασία μπορεί να προέρχονται από το RNA ανύποπτων ανθρώπινων δειγμάτων.
Λέξεις κλειδιά
SARS-CoV-2, COVID-19, Ιός, De novo Assembly, Whole genome sequencing, WGS, Bioinformatics, PCR, SARS-CoV, Bat SARS-CoV, Human immunodeficiency virus, HIV, Hepatitis virus, Measles virus, Zika virus, Ebola virus, Marburg virus.
Εισαγωγή
Για την κατασκευή αλληλουχιών ιικού γονιδιώματος, τα νουκλεϊκά οξέα (RNA ή DNA) απομονώνονται από διάφορες πηγές νουκλεϊκών οξέων, όπως υγρό βρογχοκυψελιδικού καθαρισμού (BALF) [1, 2], ρινοφαρυγγικά επιχρίσματα [3, 4, 5, 6, 12, 13], συστατικά κυτταροκαλλιέργειας ή υπερκείμενα κυτταροκαλλιέργειας [2, 11, 12, 13, 14, 16], καθώς και από δείγματα ανθρώπων [8, 9, 10, 16] και ζώων [7, 15] και αλληλουχίζονται. Κατά τη διαδικασία αυτή, τα νουκλεϊκά οξέα που λαμβάνονται δεν προέρχονται αποκλειστικά από προηγουμένως απομονωμένα σωματίδια (ιού), δηλαδή διαχωρισμένα από οτιδήποτε άλλο, αλλά συχνά από ολόκληρο το δείγμα. Έτσι, η προέλευση των τμημάτων νουκλεϊκών οξέων που χρησιμοποιούνται για τον υπολογισμό των αλληλουχιών του γονιδιώματος είναι εκ των προτέρων ασαφής.
Στην περίπτωση των ριβονουκλεϊκών οξέων (RNA), αυτά μεταγράφονται πρώτα σε cDNA με τη χρήση RNA-εξαρτώμενης DNA πολυμεράσης. Στη συνέχεια, το DNA ή το cDNA κατακερματίζεται με τη βοήθεια ενζύμων και ενισχύεται με αλυσιδωτή αντίδραση πολυμεράσης (PCR) πριν από την πραγματική αλληλούχιση, δηλαδή τον προσδιορισμό της νουκλεοτιδικής αλληλουχίας των μικρών τμημάτων DNA ή cDNA. Κατά την ενίσχυση, εκτός από τις τυχαίες αλληλουχίες εκκινητών (τυχαία εξαμερή), χρησιμοποιούνται επίσης πολύ ειδικές αλληλουχίες εκκινητών ανάλογα με τα γονιδιώματα αναφοράς ή τα γονιδιώματα-στόχους που εξετάζονται [π.χ.: 1, 3, 4, 5, 6, 7, 8, 17, 18]. Τέλος, τα δεδομένα αλληλουχίας που λαμβάνονται με τον τρόπο αυτό υποβάλλονται σε επεξεργασία με αλγόριθμους βιοπληροφορικής.
Δύο συνήθεις μέθοδοι για τον προσδιορισμό των ακολουθιών του ιικού γονιδιώματος αντιπροσωπεύουν την de novo μετα-τρανσκριπτομική συναρμολόγηση [1, 12] και την αλληλούχιση ολόκληρου του γονιδιώματος [3, 4, 5, 6, 17, 18]. Ενώ η de novo μετα-τρανσκριπτομική συναρμολόγηση συχνά δεν χρησιμοποιεί καθόλου αλληλουχίες αναφοράς ή μόνο ακολουθίες αναφοράς κατάντη, η αλληλούχιση ολόκληρου γονιδιώματος χρησιμοποιεί μεγάλο αριθμό ειδικών αλληλουχιών εκκινητών, ορισμένες από τις οποίες καλύπτουν ήδη από κοινού το 4% έως 17% του γονιδιώματος-στόχου [1, 17]. Για την ενίσχυση του cDNA χρησιμοποιούνται συχνά 35 έως 45 κύκλοι [1, 6, 17].
Στην περίπτωση του SARS-CoV-2 (GenBank: MN908947.3) [1], η πρόταση αλληλουχίας του ιικού γονιδιώματος υπολογίστηκε με de novo μετα-τρανσκριπτομική συναρμολόγηση ολικού RNA από το BALF ενός ασθενούς στο Wuhan της Κίνας. Για τη συναρμολόγηση των contigs χρησιμοποιήθηκαν οι assemblers Megahit (v.1.1.3) και Trinity (v.2.5.1). Το Megahit δημιούργησε συνολικά 384.096 (200 nt - 30.474 nt) και το Trinity υπολόγισε 1.329.960 (201 nt - 11.760 nt) contigs. Οι μεγάλες διαφορές μεταξύ των δύο συναρμολογήσεων είναι αξιοσημείωτες. Σύμφωνα με το [1], το μεγαλύτερο contig που συγκεντρώθηκε με το Megahit παρουσίασε υψηλή νουκλεοτιδική ομοιότητα (89,1%) με το γονιδίωμα της νυχτερίδας SL-CoVZC45 (GenBank: MG772933) και χρησιμοποιήθηκε για το σχεδιασμό εκκινητών για την επιβεβαίωση της PCR και των τερματικών του γονιδιώματος.
Η οργάνωση του γονιδιώματος του ιού προσδιορίστηκε με ευθυγράμμιση ακολουθιών με δύο αντιπροσωπευτικά είδη του γένους Betacoronavirus, έναν κορονoϊό που σχετίζεται με τον άνθρωπο (SARS-CoV Tor 2, GenBank: AY274119) και έναν κορονοϊό που σχετίζεται με τη νυχτερίδα (νυχτερίδα SL- CoVZC45, GenBank: MG772933).
Από το δείγμα του ασθενούς δεν ταυτοποιήθηκε και δεν χαρακτηρίστηκε βιοχημικά κανένα παθογόνο ιϊκό σωματίδιο που να σχετίζεται μοναδικά με την αλληλουχία MN908947.3. Αντίθετα, έγινε εξαγωγή και επεξεργασία ολικού RNA από το BALF ενός ασθενούς. Δεν υπάρχουν στοιχεία που να αποδεικνύουν ότι χρησιμοποιήθηκαν μόνο ιϊκά νουκλεϊκά οξέα για την κατασκευή του ισχυριζόμενου ιϊκού γονιδιώματος για τον SARS-CoV-2. Περαιτέρω, όσον αφορά την κατασκευή της ισχυριζόμενης αλυσίδας ιϊκού γονιδιώματος, δεν έχουν δημοσιευθεί αποτελέσματα πιθανών πειραμάτων ελέγχου. Αυτό ισχύει εξίσου για όλες τις άλλες αλληλουχίες αναφοράς που εξετάζονται στην παρούσα εργασία. Στην περίπτωση του SARS-CoV-2, ένας προφανής έλεγχος θα ήταν ότι το διεκδικούμενο ιϊκό γονιδίωμα δεν μπορεί να συναρμολογηθεί από μη υποψιασμένες πηγές RNA ανθρώπινης ή ακόμη και άλλης προέλευσης.
Στην παρούσα δημοσίευση, διερευνήσαμε την αναπαραγωγιμότητα των de novo συναρμολογήσεων χρησιμοποιώντας τα αρχικά δημοσιευμένα δεδομένα αλληλουχίας για την αρχική εργασία σχετικά με τον κορονoϊό SARS- CoV-2 [1]. Διερευνήσαμε περαιτέρω τη δομική ομοιότητα των παρόντων δεδομένων αλληλουχίας με άλλες δημόσια διαθέσιμες ιϊκές αλληλουχίες αναφοράς για τον ιό (νυχτερίδα) SARS-CoV [1, 7, 13, 14], τον ιό της ανθρώπινης ανοσοανεπάρκειας [8], τον ιό της δέλτα ηπατίτιδας [9], τον ιό της ιλαράς [11, 12], τον ιό Zika [10], τον ιό Ebola [15] και τον ιό Marburg [16] (Πίνακες και Σχήματα: Πίνακας 3). Για τον σκοπό αυτό, παρουσιάζουμε εδώ ένα απλό πρωτόκολλο βιοπληροφορικής. Για να επικυρώσουμε τα αποτελέσματά μας, εξετάσαμε επίσης τυχαία παραγόμενες και φανταστικές αλληλουχίες γονιδιώματος για να αποκλείσουμε την καθαρή τυχαιότητα στα αποτελέσματά μας.
Κύρια ενότητα
Ανανεωμένη de novo συναρμολόγηση δημοσιευμένων δεδομένων ακολουθίας
Για να επαναλάβουμε τη συναρμολόγηση de novo, κατεβάσαμε τα αρχικά δεδομένα αλληλουχίας (SRR10971381) από τις 27/01/2020 στις 30/11/2021 χρησιμοποιώντας τα εργαλεία SRA [19] από το Διαδίκτυο. Για να προετοιμάσουμε τις ανάγνωσης ανά ζεύγη για το στάδιο της πραγματικής συναρμολόγησης με το Megahit (v.1.2.9) [20], χρησιμοποιήσαμε τον προεπεξεργαστή FASTQ fastp (v.0.23.1) [21]. Μετά το φιλτράρισμα των αναγνώσεων ζευγών άκρων, παρέμειναν 26.108.482 από το αρχικό σύνολο των 56.565.928 αναγνώσεων, με μήκος περίπου 150 bp. Ένα μεγάλο ποσοστό των αλληλουχιών, πιθανότατα η πλειονότητα των αλληλουχιών ανθρώπινης προέλευσης, αντικαταστάθηκε από τους συγγραφείς με «N» για άγνωστο και επομένως φιλτράρεται από το fastp. Αυτό πρέπει να θεωρηθεί προβληματικό με την έννοια της επιστημονικότητας, δεδομένου ότι δεν μπορούν να ανακαλυφθούν ή να αναπαραχθούν όλα τα βήματα. Για την περίτεχνη δημιουργία περιγράμματος από τις υπόλοιπες σύντομες αναγνώσεις ακολουθίας, χρησιμοποιήσαμε το Megahit (v.1.2.9) χρησιμοποιώντας την προεπιλεγμένη ρύθμιση.
Λάβαμε 28.459 (200 nt - 29.802 nt) contigs, σημαντικά λιγότερα από όσα περιγράφονται στο [1]. Σε απόκλιση από τις αναπαραστάσεις στο [1], το μεγαλύτερο contig που συγκεντρώσαμε περιλάμβανε μόνο 29.802 nt, 672 nt λιγότερο από το μεγαλύτερο contig με 30.474 nt, το οποίο σύμφωνα με το [1] περιλάμβανε σχεδόν ολόκληρο το ιικό γονιδίωμα. Το μακρύτερο contig μας παρουσίασε τέλεια ταύτιση με την αλληλουχία MN908947.3 σε μήκος 29.801 nt (Πίνακες και Σχήματα, Πίνακες 1, 2). Έτσι, δεν μπορέσαμε να αναπαραγάγουμε το μακρύτερο contig των 30.474 nt, το οποίο είναι τόσο σημαντικό για την επιστημονική επαλήθευση. Κατά συνέπεια, τα δημοσιευμένα δεδομένα αλληλουχίας δεν μπορούν να είναι οι αρχικές αναγνώσεις που χρησιμοποιήθηκαν για τη συναρμολόγηση.
Μετά τη συναρμολόγηση των contigs, προσδιορίσαμε τον αντίστοιχο πλούτο κάλυψης με την αντιστοίχιση των σύντομων αλληλουχιών στα 28.459 προσδιορισμένα contigs χρησιμοποιώντας το Bowtie2 (v.2.4.4) [22]. Στη συνέχεια αντιστοιχίσαμε τα 50 contigs με την υψηλότερη αφθονία κάλυψης και τα 50 μεγαλύτερα contigs με τη βάση δεδομένων νουκλεοτιδίων (Blastn) στις 12/05/2021 και στις 12/20/2021, αντίστοιχα. Τα λεπτομερή αποτελέσματα της αναζήτησης μπορείτε να τα βρείτε στους πίνακες και τις εικόνες: Πίνακες 1, 2.
Σύγκριση των αποτελεσμάτων μας (Πίνακες και Σχήματα: Πίνακας 1) με εκείνα του [1, Συμπληρωματικός Πίνακας 1. Τα 50 κορυφαία άφθονα συναρμολογημένα contigs που δημιουργήθηκαν με το πρόγραμμα Megahit.] παρουσιάζουν αξιοσημείωτες διαφορές. Στη συνέχεια, τα αναγνωριστικά contig από το [1] έχουν μπροστά τους το «1_» για να διακρίνονται καλύτερα από τα δικά μας αναγνωριστικά contig. Σε γενικές γραμμές, μπορεί να δηλωθεί ότι τα αποτελέσματα της ερώτησής μας σχετικά με τους αριθμούς προσθήκης δεν ταιριάζουν ακριβώς με εκείνα του [1]. Όσον αφορά τις περιγραφές των θεμάτων, παρατηρήσαμε καλή ταύτιση ως επί το πλείστον. Περαιτέρω, με εξαίρεση το μεγαλύτερο σε μήκος contig (1_k141_275316), τα δικά μας contigs βρέθηκαν να έχουν μεγαλύτερο μήκος και τείνουν να έχουν μεγαλύτερο πλούτο κάλυψης. Η περίπτωση είναι σαφής για το contig 1_k141_179411 σε σύγκριση με το contig k141_12253. Το πρώτο έχει μήκος 2.733 nt, ενώ το δεύτερο έχει μήκος 5.414 nt. Αυτό παρέχει την πρώτη πιθανή ένδειξη ότι κατά την επιβεβαίωση με PCR με εκκινητές που κατασκευάστηκαν για τον MN908947.3 από το 1_k141_275316 (30,474 nt) σημειώθηκε μη ειδική ενίσχυση αναγνώσεων αλληλουχίας που δεν σχετίζονται με τον SARS-CoV-2.
Σε αυτό το σημείο, θα πρέπει να συζητηθεί λεπτομερώς το contig με την ταυτοποίηση k141_27232, με το οποίο συνδέονται 1.407.705 αλληλουχίες, και συνεπώς περίπου το 5% των υπόλοιπων 26.108.482 αλληλουχιών. Η ευθυγράμμιση με τη βάση δεδομένων νουκλεοτιδίων στις 05/12/2021 έδειξε υψηλή ταύτιση (98,85%) με το «Homo sapiens RNA, 45S pre- ribosomal N4 (RNA45SN4), ribosomal RNA» (GenBank: NR_146117.1, με ημερομηνία 04/07/2020). Η παρατήρηση αυτή έρχεται σε αντίθεση με τον ισχυρισμό στο [1] ότι έγινε απομάκρυνση του ριβοσωματικού RNA και ότι οι αναγνώσεις ανθρώπινης αλληλουχίας φιλτράρονταν με τη χρήση του ανθρώπινου γονιδιώματος αναφοράς (human release 32, GRCh38.p13). Ιδιαίτερη σημείωση εδώ είναι το γεγονός ότι η αλληλουχία NR_146117.1 δεν δημοσιεύθηκε παρά μόνο μετά τη δημοσίευση της βιβλιοθήκης αλληλουχιών SRR10971381 που εξετάζεται εδώ.
Η παρατήρηση αυτή υπογραμμίζει τη δυσκολία να προσδιοριστεί εκ των προτέρων η ακριβής προέλευση των επιμέρους τμημάτων νουκλεϊκών οξέων που χρησιμοποιούνται για την κατασκευή των ακολουθιών του ιϊκού γονιδιώματος.
Ανάλυση της δομής της αλληλουχίας βάσει αναφοράς
Βασικά, αντιστοιχίσαμε τις ανάγνωσης ανά ζεύγη (2x151 bp) με το BBMap [23] στις αλληλουχίες αναφοράς που εξετάσαμε (Πίνακες και Σχήματα: Πίνακας 3) χρησιμοποιώντας σχετικά μη ειδικές ρυθμίσεις. Στη συνέχεια μεταβάλαμε το ελάχιστο μήκος (M1) και την ελάχιστη (νουκλεοτιδική) ταυτότητα (M2) με το reformat.sh για να λάβουμε αντίστοιχα υποσύνολα των προηγουμένως χαρτογραφημένων αλληλουχιών με κατάλληλη ποιότητα. Η αύξηση του ελάχιστου μήκους M1 ή της ελάχιστης νουκλεοτιδικής ταυτότητας M2 αυξάνει έτσι τη σημασία της αντίστοιχης χαρτογράφησης. Στη συνέχεια, σχηματίσαμε αλληλουχίες συναίνεσης με τα αντίστοιχα υποσύνολα επιλεγμένης ποιότητας σε σχέση με την επιλεγμένη αναφορά. Θέσαμε όλες τις βάσεις με ποιότητα μικρότερη από 20 σε «Ν» (άγνωστο). Ποιότητα 20 σημαίνει ποσοστό σφάλματος 1% ανά νουκλεοτίδιο, το οποίο μπορεί να θεωρηθεί επαρκές στο πλαίσιο των αναλύσεών μας. Τέλος, η αξιολόγηση της συμφωνίας μεταξύ των αλληλουχιών αναφοράς και συναίνεσης πραγματοποιήθηκε με τη χρήση των BWA [24], Samtools [25] και Tablet [26]. Το διατεταγμένο ζεύγος (M1; M2) = (37; 0,6) επιλέχθηκε ακριβώς για να δώσει ποσοστά σφάλματος F1 και F2, αντίστοιχα, μικρότερα από 10% για την αναφορά LC312715.1. Τα αποτελέσματα όλων των υπολογισμών που πραγματοποιήθηκαν παρουσιάζονται στους πίνακες και στα σχήματα: Πίνακας 4.
Οι υπολογισμοί δείχνουν την υψηλότερη σημασία για την επιλογή του διατεταγμένου ζεύγους (37; 0,6), η οποία μπορεί να φανεί από τα υψηλότερα ποσοστά σφάλματος σε κάθε περίπτωση. Συγκρίσιμη σημασία παρέχουν τα διατεταγμένα ζεύγη (47; 0,50) και (25; 0,62). Ενώ οι αλληλουχίες γονιδιωμάτων που σχετίζονται με τους κορονοϊούς παρουσιάζουν ποσοστά σφάλματος περίπου άνω του 10% για όλα τα εξεταζόμενα διατεταγμένα ζεύγη (M1; M2), τα ποσοστά σφάλματος των δύο αλληλουχιών LC312715.1 (HIV) και NC_001653.2 (δέλτα ηπατίτιδας) είναι κάτω του 10% και μειώνονται περαιτέρω για τα διατεταγμένα ζεύγη (32; 0,60) και (30; 0,60). Η αλληλουχία MG772933_short αποτελείται κυρίως από το τμήμα που δεν μπορεί να καλυφθεί με τις αναγνώσεις που σχετίζονται με τον SARS-CoV-2 (βλέπε πίνακες και σχήματα: σχήμα 3). Και πάλι, δεν επιτεύχθηκε βελτίωση με τη μείωση των τιμών για τα M1 και M2. Τα ποσοστά σφάλματος για τις αλληλουχίες NC_039345.1 (ιός Ebola), NC_024781.1 (ιός Marburg), AF266291.1 και KJ410048.1 (ιός ιλαράς) είναι σημαντικά υψηλότερα από εκείνα των LC312715.1 και NC_001653.2. Ενώ οι αλληλουχίες νουκλεϊκών οξέων που χρησιμοποιήθηκαν για τον υπολογισμό των πρώτων γονιδιωμάτων πολλαπλασιάστηκαν σε κύτταρα Vero, οι αλληλουχίες νουκλεϊκών οξέων που χρησιμοποιήθηκαν για τους LC312715.1 και NC_001653.2 προήλθαν απευθείας από δείγματα ανθρώπινης προέλευσης (πίνακες και σχήματα: Πίνακας 3). Επομένως, τίθεται το ερώτημα αν το αποτέλεσμα αυτό οφείλεται σε δομικές διαφορές των αντίστοιχων πηγών νουκλεϊκών οξέων ή στα αντίστοιχα πρωτόκολλα αλληλούχισης που χρησιμοποιήθηκαν. Για παράδειγμα, η αντίστροφη μεταγραφάση που χρησιμοποιείται για τη μετατροπή του RNA σε cDNA ή οι αλληλουχίες εκκινητών που χρησιμοποιούνται για την ενίσχυση καθώς και οι κύκλοι ενίσχυσης θα μπορούσαν ενδεχομένως να οδηγήσουν σε διαφορές στις βιβλιοθήκες αλληλουχιών που λαμβάνονται.
Τα υψηλότερα ποσοστά σφάλματος F1 και F2 παρουσιάζονται από τις τυχαία παραγόμενες πλασματικές αλληλουχίες γονιδιώματος rnd_uniform, rnd_wuhan, rnd_wh_mk_1 και rnd_wh_mk_2, επομένως τα αποτελέσματα που βρέθηκαν εδώ δεν είναι καθαρά τυχαία.
Γραφική ανάλυση των κατανομών κάλυψης και των μηκών ανάγνωσης
Αφού παρατηρήσαμε τη δυνατότητα σχηματισμού αλληλουχιών συναίνεσης με υψηλή ποιότητα σε σχέση με ορισμένες αλληλουχίες αναφοράς, αναλύσαμε την κατανομή της κάλυψης των σχετικών αναγνώσεων βραχείας αλληλουχίας (πίνακες και σχήματα: σχήματα 1-22) και την κατανομή των μηκών ανάγνωσης (πίνακες και σχήματα: σχήματα 23-25). Για τον σκοπό αυτό, χαρτογραφήσαμε προηγουμένως τις αναγνώσεις βραχείας αλληλουχίας στις αντίστοιχες αλληλουχίες αναφοράς τους χρησιμοποιώντας BBMap, ((M1; M2) = (37; 0,60)). Εκτός από τις σύντομες αλληλουχίες, αντιστοιχίσαμε επίσης τα 26 ζεύγη εκκινητών [1, Συμπληρωματικός πίνακας 8. Εκκινητές PCR που χρησιμοποιήθηκαν στην παρούσα μελέτη.] για την αλληλούχιση ολόκληρου του γονιδιώματος του SARS-CoV-2 (GenBank: MN908947.3) στα υπό εξέταση γονιδιώματα αναφοράς. Η επακόλουθη ανάλυση πραγματοποιήθηκε μέσω Tablet και του προγράμματος υπολογιστικών φύλλων Excel.
Πρώτον, εξετάζουμε την τυχαία παραγόμενη αναφορά rnd_uniform. Ανάλογες παρατηρήσεις ισχύουν για τα τυχαία παραγόμενα γονιδιώματα αναφοράς rnd_wuhan, rnd_wh_mk_1 και rnd_wh_mk_2 (πίνακες και σχήματα: σχήματα 14-16).
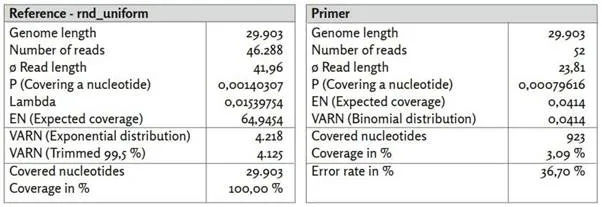
Εικόνα 13: Αναφορά rnd_uniform.
α) rnd_uniform_reads χαρτογραφημένο με χρήση BBMap, (M1; M2) = (37; 0,60).
β) rnd_uniform_primer χαρτογραφημένο με χρήση BBMap.
γ) Η εκθετική κατανεμημένη κάλυψη δημιουργήθηκε με στοχαστική προσομοίωση με τη μέθοδο της αντιστροφής.
δ) Τα 26 ζεύγη εκκινητών ([1, Συμπληρωματικός πίνακας 8. Εκκινητές PCR που χρησιμοποιήθηκαν στην παρούσα μελέτη.]) είναι άνισα κατανεμημένα σε ολόκληρο το γονιδίωμα αναφοράς. Οι θέσεις των εκκινητών συσχετίζονται μόνο ασθενώς με περιοχές υψηλής νουκλεοτιδικής κάλυψης, καθεμία από τις οποίες περιλαμβάνει μόνο μερικά νουκλεοτίδια.
ε) Η κατανομή των rnd_uniform_reads φαίνεται σε μεγάλο βαθμό τυχαία. Η διακύμανση της εξεταζόμενης εκθετικής κατανομής συμφωνεί καλά με την περικομμένη εμπειρική διακύμανση.
Η κάλυψη (rnd_uniform_reads) μεταβάλλεται τυχαία και σχετικά ομοιογενώς σε όλες τις θέσεις νουκλεοτιδίων. Η δομή είναι συγκρίσιμη με την τυχαία παραγόμενη κάλυψη (εκθετική κατανομή κάλυψης), αν και η διακύμανση εμφανίζεται κάπως χαμηλότερη. Σε μερικές απομονωμένες νουκλεοτιδικές θέσεις, η κάλυψη παρουσιάζει υψηλή κάλυψη σε σύγκριση με τον μέσο όρο, αλλά κάθε μία από αυτές καλύπτει μόνο μερικές συνεχόμενες νουκλεοτιδικές περιοχές. Η συσχέτιση με τις θέσεις των εκκινητών είναι ελάχιστα έντονη. Η αμιγώς τυχαία εμφανιζόμενη κάλυψη με τις σύντομες αναγνώσεις αλληλουχίας συσχετίζεται με μια μη συνεχή αντιστοιχήσιμη αλληλουχία συναίνεσης και υψηλό ποσοστό σφάλματος F1 38,60%. Έτσι, η τυχαία (εσωτερική) νουκλεοτιδική δομή της στοχαστικά προσομοιωμένης αλληλουχίας αναφοράς «rnd_uniform» μάλλον απουσιάζει από τα δεδομένα αλληλουχίας που εξετάζονται εδώ.
Αντίθετα, εξετάζουμε τώρα το γονιδίωμα αναφοράς για τον SARS-CoV-2 (GenBank: MN908947.3).
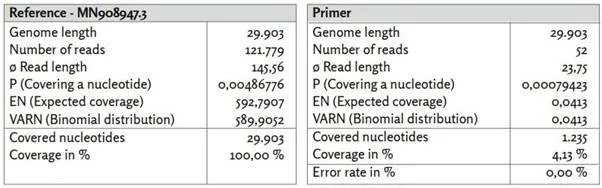
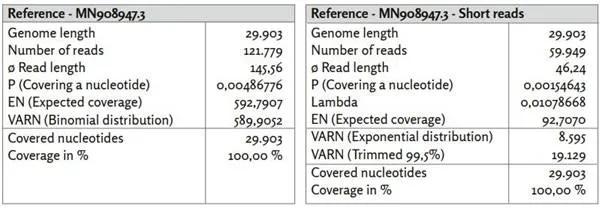
Εικόνα 1: Γονίδιο αναφοράς MN908947.3.
α) MN908947.3_reads χαρτογραφημένα με το Bowtie2 υπό χρήση προεπιλεγμένων ρυθμίσεων.
β) MN908947.3_primer χαρτογραφημένο με το BBMap.
γ) Τα ποσοστά προσδιορίστηκαν από τα EN και VARN υπό την υπόθεση κατανομής διωνυμικής κατανομής.
δ) Τα 26 ζεύγη εκκινητών ([1], Συμπληρωματικός πίνακας 8. Εκκινητές PCR που χρησιμοποιήθηκαν στην παρούσα μελέτη.) είναι ομοιόμορφα κατανεμημένα σε ολόκληρο το γονιδίωμα αναφοράς. Οι θέσεις των εκκινητών συσχετίζονται με περιοχές υψηλής νουκλεοτιδικής κάλυψης.
Σε αντίθεση με την Εικόνα 13, η κατανομή της κάλυψης παρουσιάζει περισσότερο ένα κυματοειδές μοτίβο με τακτικές σημαντικά αυξημένες καλύψεις νουκλεοτιδίων. Τα 26 ζεύγη εκκινητών κατανέμονται ομοιόμορφα σε όλες τις νουκλεοτιδικές θέσεις της αλληλουχίας αναφοράς. Οι θέσεις εκκινητών βρίσκονται συχνά κοντά σε νουκλεοτιδικές θέσεις με υψηλή νουκλεοτιδική κάλυψη σε σύγκριση με τον μέσο όρο. Αυτό δείχνει ότι δεν ενισχύθηκαν εξίσου όλα τα τμήματα του γονιδιώματος αναφοράς. Υποθέτοντας ότι και οι 29.903 νουκλεοτιδικές θέσεις είναι εξίσου πιθανό να εμφανιστούν στις σχετικές με τον SARS-CoV-2 αναγνώσεις, η κάλυψη για κάθε νουκλεοτιδική θέση θα πρέπει να βρίσκεται μεταξύ των δύο γραμμών με πιθανότητα 99,5% (υποθέτοντας διωνυμική κατανομή). Αυτό δεν ισχύει για το 90% περίπου των νουκλεοτιδικών θέσεων. Εκ των προτέρων, θα περίμενε κανείς ότι εάν υπάρχει επαρκές ιϊκό RNA στο δείγμα και διαβάζονται επαρκή κομμάτια αλληλουχίας, θα επιτυγχανόταν ομοιογενής κάλυψη των νουκλεοτιδίων εντός του ιικού γονιδιώματος.
Το ακόλουθο γράφημα επιτρέπει τη μελέτη των κατανομών των μηκών ανάγνωσης των αναφορών που μόλις εξετάστηκαν (rnd_uniform και MN908947.3)
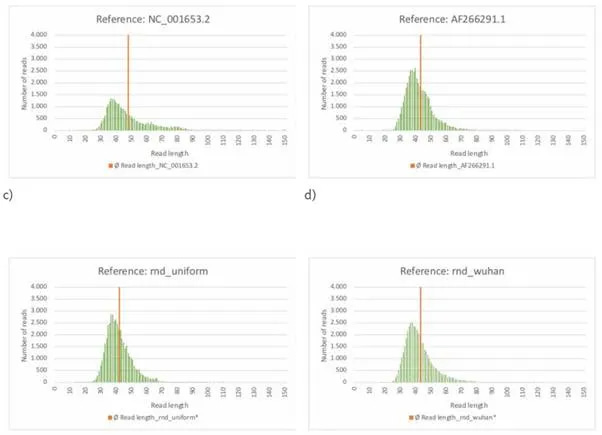
Εικόνα 23: α)-στ)
Χαρτογράφηση με BBMap (M1, M2) = (37, 0,60).
Ανάλυση στο Excel.
Στην εικόνα 23ε) παρουσιάζεται η κατανομή των μηκών ανάγνωσης στην περίπτωση της αναφοράς "rnd_uniform". Το μέσο μήκος ανάγνωσης είναι 41,96 nt, ελάχιστα δεξιά από το μέγιστο της κατανομής. Συγκριτικά, η κατανομή για την αναφορά MN908947.3, Εικόνα 23α) δείχνει μια εξέχουσα (τυχαία) περιοχή παρόμοια με την Εικόνα 23ε) και μια διακριτή περιοχή με αναγνώσματα μήκους περίπου 150 nt. Το μέσο μήκος ανάγνωσης είναι πάνω από 110 nt. Όλες οι αλληλουχίες αναφοράς με συγκρίσιμη και επομένως μάλλον τυχαία κατανομή των μηκών ανάγνωσης όπως στην στοχαστικά προσομοιωμένη αναφορά «rnd_uniform» (Πίνακες και σχήματα: Εικόνα 23δ), στ), Εικόνα 24δ), ε), στ), Εικόνα 25α) - γ)) παρουσιάζουν επίσης υψηλά ποσοστά σφάλματος F1 και F2 (Πίνακες και σχήματα: Πίνακας 4).
Η διαπίστωση αυτή υπογραμμίζεται από την ακόλουθη ανάλυση. Προκειμένου να κατανοήσουμε καλύτερα την εσωτερική δομή των δημοσιευμένων περίπου 56 εκατομμυρίων αλληλουχιών, εξετάσαμε την πρόσθετη συνθήκη maxlength=100 για την αλληλουχία MN908947.3 κατά τον σχηματισμό υποσυνόλων μετά τη χαρτογράφηση με το BBMap εκτός από τα M1 και M2.
Εικόνα 2: Αναφορά MN908947.3.
α) MN908947_reads χαρτογραφημένο με το Bowtie2 με χρήση προεπιλεγμένων ρυθμίσεων.
β) MN908947_short_reads χαρτογραφημένο με χρήση BBMap, (M1; M2) = (37 (max. 100); 0,60).
γ) Η εκθετική κατανεμημένη κάλυψη δημιουργήθηκε με στοχαστική προσομοίωση με τη μέθοδο της αντιστροφής. Η κατανομή κάλυψης MN908947_short_reads παρουσιάζει ένα πιο τυχαίο μοτίβο, αλλά έχει υψηλότερη κομμένη διακύμανση. Αυτό οφείλεται κυρίως στις λίγες διακυμάνσεις της κατανομής κάλυψης.
Με τον αποκλεισμό όλων των αντιστοιχιζόμενων αλληλουχιών μήκους άνω των 100 νουκλεοτιδίων, ουσιαστικά αφαιρέθηκαν οι περίπου 120.000 αναγνώσεις που σχετίζονται με τον SARS-CoV-2. Η κατανομή της κάλυψης των εναπομεινάντων μικρών αλληλουχιών εμφανίζεται πλέον τυχαία, κατ' αναλογία με την Εικόνα 13. Και πάλι, αυτό συσχετίζεται με τα υψηλά ποσοστά σφάλματος R1 (29,90%) και R2 (29,96%). Αυτό δείχνει ότι καμία σημαντική δομή της αναφοράς MN908947.3 δεν περιλαμβάνεται στις δημοσιευμένες αλληλουχίες, εκτός από τις περίπου 120.000 (Πίνακες και Σχήματα. Πίνακας 1) σχετικές σύντομες αναγνώσεις.
Πριν αναφερθούμε λεπτομερώς σε ορισμένα από τα γονιδιώματα αναφοράς που εξετάσαμε, θα θέλαμε πρώτα να εξετάσουμε την κάλυψη δύο άλλων contigs k141_12253 και k141_20796. Ενώ το contig που αναγνωρίστηκε ως k141_12253 χαρακτηρίζεται από σχετικά υψηλή κάλυψη, το k141_20796 είναι μεταξύ των τριών μεγαλύτερων contigs που υπολογίστηκαν.
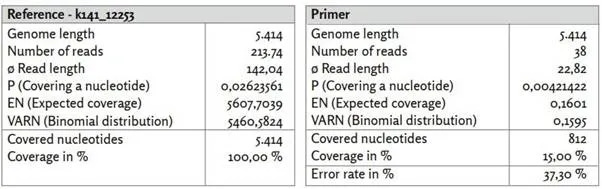
Εικόνα 18: Αναφορά k141_12253.
α) k141_12253_reads χαρτογραφημένα με το Bowtie2 με χρήση προεπιλεγμένων ρυθμίσεων.
β) k141_12253_primer χαρτογραφημένο με χρήση του BBMap.
Το contig k141_12253 παρουσιάζει υψηλή ομοιότητα με το βακτήριο Leptotrichia (GenBank: CP012410.1). Από τις 52 δημοσιευμένες αλληλουχίες εκκινητών, οι 38 μπόρεσαν να χαρτογραφηθούν στο σημείο αναφοράς k141_12253 με σχετικά υψηλό ποσοστό σφάλματος 37,30%. Η κατανομή της κάλυψης αποδεικνύεται εξαιρετικά ανομοιογενής και παρουσιάζει, ιδίως εντός των πρώτων 500 νουκλεοτιδίων, εξαιρετικά υψηλή κάλυψη νουκλεοτιδίων σε σύγκριση με τον μέσο όρο. Οι περιοχές με υψηλή κάλυψη συσχετίζονται με τις καθορισμένες θέσεις των εκκινητών. Αυτό θα μπορούσε να υποδηλώνει ότι δεν ενισχύθηκαν σε μεγάλες ποσότητες αποκλειστικά αναγνώσεις που σχετίζονται με τον SARS-CoV-2. Λαμβάνοντας υπόψη το σχετικά υψηλό ποσοστό σφάλματος 37,30%, αυτό θα σήμαινε μια σχετικά μη ειδική ενίσχυση. Έτσι, τίθεται το ερώτημα αν οι αναγνώσεις που προέκυψαν από την ενίσχυση του cDNA με τις συγκεκριμένες αλληλουχίες εκκινητών ήταν ήδη παρούσες στο αρχικό δείγμα ή δημιουργήθηκαν από την ίδια τη διαδικασία.
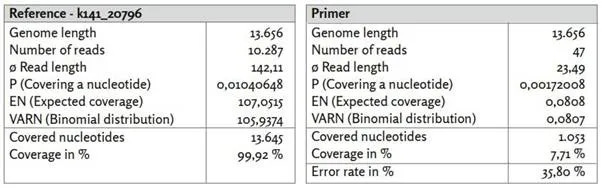
Εικόνα 21: Αναφορά k141_20796.
α) k141_20796_reads χαρτογραφημένα με το Bowtie2 με χρήση προεπιλεγμένων ρυθμίσεων.
β) k141_20796_primer χαρτογραφημένο με χρήση του BBMap.
Το contig k141_20796, το οποίο έχει υψηλή ταύτιση με το βακτήριο Veillonella parvula (GenBank: LR778174.1), εμφανίζει χαμηλότερη κάλυψη με συσχετιζόμενες αναγνώσεις σε σύγκριση με το contig με ταυτοποίηση k141_12253. Η δομή της νουκλεοτιδικής κάλυψης είναι παρόμοια με εκείνη του SARS-CoV-2 (GenBank: MN908947.3). Ειδικότερα, η κάλυψη είναι και πάλι ανομοιογενής, υποδηλώνοντας ανομοιογενή ενίσχυση. Λόγω του μεγαλύτερου μήκους νουκλεοτιδίων, 47 από τις 52 δημοσιευμένες αλληλουχίες εκκινητών μπορούσαν τώρα να αντιστοιχιστούν στο κονδύλιο αναφοράς με μέσο ποσοστό σφάλματος 35,80%. Και πάλι, οι θέσεις των εκκινητών συσχετίζονται καλά με περιοχές υψηλής νουκλεοτιδικής κάλυψης. Αυτό θα μπορούσε και πάλι να υποδηλώνει μη ειδική ενίσχυση αλληλουχιών που δεν σχετίζονται με τον SARS-CoV-2 (GenBank: MN908947.3).
Στην παρούσα ενότητα, θα συζητήσουμε λεπτομερέστερα τις αλληλουχίες αναφοράς «Human immunodeficiency virus 1» (GenBank: LC312715.1) και «Measles virus genotype D8 strain MVi/Muenchen» (GenBank: KJ410048.1). Όλα τα άλλα σχήματα μπορείτε να τα βρείτε στα συμπληρωματικά υλικά (πίνακες και σχήματα: σχήματα 1-22 και σχήματα 23-25).
Εικόνα 6: Αναφορά LC312715.1.
α) LC312715.1_short_reads χαρτογραφημένο με χρήση BBMap, (M1; M2) = (37; 0,60).
β) LC312715.1_primer χαρτογραφημένο με χρήση BBMap.
Ήδη στην προηγούμενη ενότητα, φάνηκε υψηλή δομική ομοιότητα των δημοσιευμένων αλληλουχιών με την αλληλουχία αναφοράς LC312715.1. Η υπολογιζόμενη αλληλουχία συναίνεσης παρουσίασε σχετικά χαμηλότερα ποσοστά σφάλματος R1 = 8,60% και R2 = 8,83% σε σύγκριση π.χ. με τις αναφορές που σχετίζονται με το SARS. Το Σχήμα 6 παρουσιάζει σαφείς διαφορές σε σχέση με το Σχήμα 13. Η κατανομή της κάλυψης παρουσιάζει επίσης περισσότερο ένα κυματοειδές μοτίβο με σχετικά κανονικές περιοχές ιδιαίτερα υψηλής κάλυψης και επομένως διαφέρει σαφώς από την κατανομή της κάλυψης της τυχαίας αναφοράς «rnd_uniform». Η κατανομή των μηκών ανάγνωσης (Εικόνα 23β), συγκρίνετε επίσης γ)) διαφέρει επίσης σημαντικά από τις πιο τυχαίες κατανομές και παρουσιάζει σημαντικό αριθμό αντιστοιχιζόμενων αναγνώσεων με μήκη μέχρι περίπου 110 nt. Το μέσο μήκος ανάγνωσης των 51,84 nt είναι επίσης υψηλότερο από ό,τι για παράδειγμα για το «rnd_uniform».
Και πάλι, είναι ενδιαφέρον να σημειωθεί η θέση των αλληλουχιών των εκκινητών σε σχέση με τις περιοχές υψηλής νουκλεοτιδικής κάλυψης σε σύγκριση με τη μεσαία κάλυψη. Συνολικά 46 από τις 52 αλληλουχίες εκκινητών μπόρεσαν να αντιστοιχιστούν στην αναφορά που εξετάζεται εδώ με ποσοστό σφάλματος 38,00%. Η εικόνα 6 υποδηλώνει ότι οι σύντομες αλληλουχίες που σχετίζονται με την αναφορά LC312715.1 ενισχύθηκαν επίσης κατά την επιβεβαίωση της PCR, παρά το γεγονός ότι οι αλληλουχίες εκκινητών μπορούσαν να αντιστοιχιστούν στην αναφορά μόνο με σχετικά υψηλό ποσοστό σφάλματος.
Τέλος, ας στραφούμε στην αναφορά KJ410048.1 (ιός της ιλαράς).
Εικόνα 10: Αναφορά KJ410048.1.
α) KJ410048.1_short_reads χαρτογραφημένο με τη χρήση BBMap, (M1; M2) = (37; 0,60).
β) KJ410048.1_primer χαρτογραφημένο με χρήση BBMap.
Η κατανομή της κάλυψης διαφέρει σημαντικά από εκείνη της εικόνας 6 και παρουσιάζει ορισμένες ομοιότητες με την κατανομή των σχετικών αναγνώσεων αλληλουχίας για το «rnd_uniform», με μικρότερη διακύμανση στις περιοχές χαμηλότερης κάλυψης. Η κατανομή των μηκών ανάγνωσης (Πίνακες και Σχήματα: Εικόνα 24d)) καθώς και το μέσο μήκος ανάγνωσης 42,38 είναι συγκρίσιμα με τα δεδομένα του «rnd_unifom» και συσχετίζονται επίσης με σχετικά υψηλά ποσοστά σφάλματος F1=28,70% και F2=28,79%.
Συζήτηση και προοπτικές
Εξετάσαμε τα δημοσιευμένα δεδομένα αλληλουχίας (αριθμός πρόσβασης BioProject PRJNA603194 στη βάση δεδομένων NCBI Sequence Read Archive (SRA)) για την αλληλουχία του γονιδιώματος του SARS-CoV-2 (GenBank: MN908947.3) χρησιμοποιώντας μια απλή προσέγγιση βιοπληροφορικής. Οι μέθοδοι που χρησιμοποιήσαμε δεν είναι ειδικές για τον SARS-CoV-2 και μπορούν να εφαρμοστούν σε άλλα δεδομένα αλληλουχίας χωρίς ειδικές τροποποιήσεις.
Πρώτον, επαναλάβαμε τη δημιουργία contig με το Megahit (v.1.2.9) χρησιμοποιώντας τα διαθέσιμα δεδομένα αλληλουχίας και λάβαμε σημαντικά διαφορετικά αποτελέσματα σε σύγκριση με τις αναπαραστάσεις στο [1]. Συγκεκριμένα, δεν μπορέσαμε να αναπαραγάγουμε το μεγαλύτερο contig μήκους 30.474 nt, το οποίο σύμφωνα με το [1] περιελάμβανε σχεδόν ολόκληρο το ιικό γονιδίωμα και λειτούργησε ως βάση για το σχεδιασμό των εκκινητών. Αντιθέτως, το μεγαλύτερο contig που δημιουργήσαμε (29.802 nt) παρουσίασε σχεδόν πλήρη ταύτιση με το MN908947.3 αναφοράς. Κατά συνέπεια, τα δημοσιευμένα δεδομένα αλληλουχίας δεν μπορεί να είναι τα αρχικά σύντομα reads που χρησιμοποιήθηκαν για τη δημιουργία contig. Αυτό πρέπει να θεωρηθεί εξαιρετικά προβληματικό στο πλαίσιο επιστημονικών δημοσιεύσεων, δεδομένου ότι με αυτόν τον τρόπο δεν είναι πλέον δυνατή η επαλήθευση των δημοσιευμένων αποτελεσμάτων. Η δυνατότητα επαλήθευσης των δημοσιευμένων επιστημονικών υποθέσεων αποτελεί την ουσία της ζωντανής επιστήμης.
Σε αντίθεση με ό,τι αναφέρθηκε στο [1], ενδέχεται να έχουμε βρει contigs με υψηλή κάλυψη που σχετίζονται με (ριβοσωμικά) ριβονουκλεϊκά οξέα ανθρώπινης προέλευσης. Έτσι, είναι πιθανό να μην εξαλείφθηκαν όλα τα νουκλεϊκά οξέα που σχετίζονται με τον άνθρωπο κατά την κατασκευή του SARS-CoV-2. Περαιτέρω, δεν παρασχέθηκαν στοιχεία για την παρουσία ιικών νουκλεϊκών οξέων στο δείγμα του ασθενούς και, κατά συνέπεια, υπάρχει πιθανότητα να χρησιμοποιήθηκαν τμήματα ανθρώπινων ή μη ιϊκών νουκλεϊκών οξέων για την κατασκευή της διεκδικούμενης ιικής αλληλουχίας MN908947.3 σε σημαντικό βαθμό χωρίς ανίχνευση. Η πιθανότητα αυτή θα πρέπει να αποκλειστεί με πειράματα ελέγχου.
Σε όλες τις δημοσιεύσεις σχετικά με τα γονιδιώματα αναφοράς που αναλύθηκαν στην παρούσα μελέτη, δεν παρασχέθηκαν επίσης τα απαραίτητα στοιχεία για την ακριβή προέλευση των τμημάτων της αλληλουχίας που χρησιμοποιήθηκαν για την κατασκευή και δεν δημοσιεύθηκαν τα απαραίτητα πειράματα ελέγχου.
Θα θέλαμε να αναφέρουμε εδώ ότι πειράματα ελέγχου μπορεί να έχουν ήδη πραγματοποιηθεί πολλές φορές χωρίς να έχουν γίνει αντιληπτά, γεγονός που δείχνει τη δυνατότητα κατασκευής γονιδιωμάτων SARS-CoV-2 από μη μολυσματικά ανθρώπινα δείγματα. Για παράδειγμα, η αλληλούχιση ολόκληρου του γονιδιώματος από δείγματα με βασική τιμή Ct μεγαλύτερη από 35 αναφέρεται στα [5] και [17]. Αυτό θα μπορούσε να αποτελέσει διάψευση του ιϊκού μοντέλου για τον SARS-CoV-2.
Η ανάλυση των κατανομών της νουκλεοτιδικής κάλυψης καθώς και των κατανομών μήκους των αντιστοιχιζόμενων αναγνώσεων αλληλουχίας για τις αντίστοιχες αλληλουχίες αναφοράς οδηγεί στην υπόθεση μιας πιθανής ακούσιας ενίσχυσης αναγνώσεων αλληλουχίας που δεν σχετίζονται με τον SARS-CoV-2. Επιπλέον, παράλληλα με αυτό, πρέπει να ληφθεί υπόψη η πιθανότητα τυχαίας δημιουργίας αλληλουχιών που δεν υπήρχαν στο αρχικό δείγμα αλλά δημιουργήθηκαν μόνο από τις συνθήκες ενίσχυσης, όπως οι αλληλουχίες εκκινητών που χρησιμοποιήθηκαν και οι κύκλοι που εκτελέστηκαν. Το ενδεχόμενο αυτό απαιτεί, επομένως, τη διενέργεια κατάλληλων πειραμάτων ελέγχου.
Εκτός από την προσπάθεια αναπαραγωγής της συναρμολόγησης που δημοσιεύθηκε στο [1] με τις δημοσιευμένες αναγνώσεις ακολουθίας, εξετάσαμε μια απλή προσέγγιση για την ανάλυση της εσωτερικής δομής μεγάλων συνόλων δεδομένων με αναγνώσεις σύντομων ακολουθιών. Με τα δεδομένα αλληλουχιών που είχαμε στη διάθεσή μας, μπορέσαμε να υπολογίσουμε αλληλουχίες συναίνεσης για τα γονιδιώματα αναφοράς LC312715.1 (HIV) και NC_001653.2 (ιός της δέλτα της ηπατίτιδας) με μεγαλύτερη αξιοπιστία από ό,τι για τις αλληλουχίες αναφοράς που θεωρήσαμε ότι σχετίζονται με τους κορονοϊούς. Αυτό ίσχυε ιδιαίτερα για το bat-SL-CoVZC45 (GenBank: MG772933.1), το οποίο οδήγησε στην υπόθεση προέλευσης του SARS-CoV-2. Έτσι, μπορέσαμε να τεκμηριώσουμε την υπόθεσή μας ότι οι ισχυριζόμενες αλληλουχίες ιικού γονιδιώματος αποτελούν παρερμηνείες, υπό την έννοια ότι έχουν κατασκευαστεί ή κατασκευάζονται απαρατήρητες από μη ιϊκά θραύσματα νουκλεϊκών οξέων. Ειδικότερα, τα αποτελέσματά μας υπογραμμίζουν την επείγουσα ανάγκη εκτέλεσης κατάλληλων πειραμάτων ελέγχου. Για κάθε ύποπτη αλληλουχία παθογόνου ιικού γονιδιώματος, ένα προφανές πρωτόκολλο θα ήταν να επιχειρείται η συναρμολόγηση των αλληλουχιών γονιδιώματος από αντίστοιχα μη ύποπτα δείγματα με τη χρήση πανομοιότυπων πρωτοκόλλων.
Παρατηρήσαμε υψηλά ποσοστά σφάλματος R1 και R2 στα γονιδιώματα αναφοράς για την ιλαρά, τον Έμπολα ή το Marburg, όπου τα θραύσματα νουκλεϊκών οξέων που χρησιμοποιήθηκαν για την κατασκευή πολλαπλασιάστηκαν σε κύτταρα Vero. Παραμένει μέχρι στιγμής ανοιχτό ερώτημα αν αυτό οφείλεται στις ίδιες τις πηγές νουκλεϊκών οξέων ή στις συνθήκες ενίσχυσης που χρησιμοποιήθηκαν (π.χ. αλληλουχίες εκκινητών και αριθμός κύκλων) ή στα πρωτόκολλα αλληλούχισης (π.χ. τις πολυμεράσες και τις αντίστροφες μεταγραφάσες που χρησιμοποιήθηκαν).
Όσον αφορά τα αποτελέσματά μας, εκτός από τη δημοσίευση των τελικών δεδομένων αλληλουχίας που χρησιμοποιήθηκαν, συνιστούμε πάντοτε τη δημοσίευση δεδομένων αλληλουχίας που προέκυψαν μόνο από ενίσχυση με τυχαία εξαμερή και μέτριους αριθμούς κύκλων, ώστε να παρέχονται όσο το δυνατόν πιο αμερόληπτα δεδομένα για τη δομική ανάλυση.
Υλικό και μέθοδοι
Βάθος κάλυψης μιας αλληλουχίας αναφοράς με σύντομες αναγνώσεις αλληλουχίας
Έστω 𝐺 το μήκος της ακολουθίας αναφοράς, Ø𝐿 το μέσο μήκος ανάγνωσης, 𝑛 ο αριθμός των αναγνώσεων βραχέων ακολουθιών και 𝑁 το τυχαίο μέσο βάθος κάλυψης της ακολουθίας αναφοράς με τις αναγνώσεις βραχέων ακολουθιών. Στη συνέχεια,

Η έκφραση Ø𝐿/G μπορεί να θεωρηθεί ως η πιθανότητα κάλυψης ενός νουκλεοτιδίου εντός της αλληλουχίας αναφοράς με μια ανάγνωση σύντομης αλληλουχίας.
Δημιουργία τυχαίων αλληλουχιών αναφοράς
Το ακόλουθο θεώρημα επιτρέπει την προσομοίωση μιας τυχαίας μεταβλητής 𝑋 με αθροιστική συνάρτηση κατανομής 𝐹.
Θεώρημα (αρχή της αντιστροφής) [28].
Έστω 𝑈 μια τυχαία μεταβλητή ομοιόμορφα κατανεμημένη στο διάστημα (0,1). Έστω 𝑋 μια τυχαία μεταβλητή με αθροιστική συνάρτηση κατανομής 𝐹, και έστω
𝐹-1(𝑦) ≔ inf {𝑥 ∈ ℝ|𝐹(𝑥) ≥ 𝑦}.
Τότε ισχύει
𝐹-1(𝑈) ~ 𝑋.
Έστω ότι 𝑈𝑖, 𝑖 = 1, ... ,29,903 είναι ανεξάρτητα ταυτόσημες τυχαίες μεταβλητές με ισοκατανομή στο διάστημα (0,1). Έστω 𝑝𝑛𝑡, 𝑛𝑡 ∈ {𝐴, 𝑇, 𝐶, 𝐺} συμβολίζει την πιθανότητα για το νουκλεοτίδιο 𝑛𝑡. Στη συνέχεια, το νουκλεοτίδιο 𝑁𝑖, 𝑖 = 1, ... ,29,903 της τυχαία παραγόμενης ακολουθίας αναφοράς λαμβάνεται μέσω
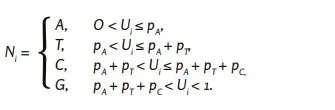
Για την ακολουθία αναφοράς «rnd_unifom» χρησιμοποιήθηκε η ομοιόμορφη κατανομή στο σύνολο {𝐴, 𝑇, 𝐶, 𝐺}. Για την προσομοίωση της τυχαίας αλληλουχίας αναφοράς «rnd_wuhan», επιλέχθηκε ως κατανομή νουκλεοτιδίων η σχετική συχνότητα εμφάνισης των νουκλεοτιδίων A, T, C και G στην αλληλουχία του γονιδιώματος του SARS-CoV-2 (GenBank: MN908947.3). Κατά την κατασκευή των τυχαιοποιημένων ακολουθιών αναφοράς «rnd_wh_mk_1» και «rnd_wh_mk_2», επιλέχθηκε η υπό συνθήκη πιθανότητα, υπό συνθήκη για το τελευταίο και για τα δύο τελευταία νουκλεοτίδια, αντίστοιχα, σύμφωνα με τις αντίστοιχες εμπειρικές συχνότητες στην ακολουθία για τον SARS-CoV-2 (GenBank: MN908947.3).
Στοχαστική προσομοίωση τυχαίων καλύψεων μιας αλληλουχίας αναφοράς
Η αθροιστική συνάρτηση κατανομής της εκθετικής κατανομής με παράμετρο 𝜆 είναι [28],
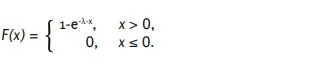
Έστω 𝑋 μια τυχαία μεταβλητή με συνάρτηση κατανομής 𝐹. Τότε ισχύει
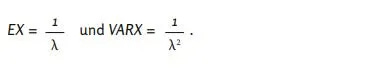
Μέθοδοι βιοπληροφορικής (δομική ανάλυση)
1. Χαρτογράφηση με χρήση BBMap
bbmap.sh ref=$reference.fasta
mapPacBio.sh in=SRR10971381_1.fastq in2=SRR10971381_2.fastq outm=mapped.sam vslow k=8 maxindel=0 minratio=0.1
2. Επιλογή των χαρτογραφημένων αλληλουχιών ανάλογα με τις M1 και M2 με χρήση του BBMap (reformat.sh)
reformat.sh in=mapped.sam out=sample_selection.sam minlength=$M1 (maxlength=100) idfilter=$M2 ow=t
3. Υπολογισμός της αλληλουχίας συναίνεσης
3.1. Προετοιμασία με τη χρήση του Samtools
samtools view -b sample_selection.sam > sample.bam samtools sort sample.bam -o sample_sort_reads.bam samtools index sample_sort_reads.bam
3.2 Προσδιορισμός της προκαταρκτικής αλληλουχίας συναίνεσης
samtools mpileup -uf mapping/$reference.fasta sample_sort_reads.bam | bcftools call -c | vcfutils.pl vcf2fq > SAMPLE_cns.fastq
3.3 Προσδιορισμός της τελικής αλληλουχίας συναίνεσης (τουλάχιστον Q20)
seqtk seq -aQ64 -q20 -n N sample_cns.fastq > sample_cns.fasta
4. Χαρτογράφηση της αλληλουχίας συναίνεσης στην αλληλουχία αναφοράς με τη χρήση BWA.
bwa index $reference.fasta
bwa mem $reference.fasta sample_cns.fasta > sample_cns.sam
5. Επανεξέταση με Tablet και Excel
Η αξιολόγηση πραγματοποιήθηκε με τη χρήση του λογισμικού Tablet για την οπτικοποίηση των δεδομένων αλληλουχίας και του προγράμματος υπολογιστικών φύλλων Excel.
Αναφορές:
s
Fan Wu u. a. A new coronavirus associated with human respiratory disease in China. In: Nature 580.7803 (2020). DOI: 10.1038/s41586-020-2202-3.
Na Zhu u. a. A Novel Coronavirus from Patients with Pneumonia in China, 2019. In: New England Journal of Medicine 382.8 (2020), S. 727-733. DOI:10.1056/nejmoa2001017.
Divinlal Harilal u. a. SARS-CoV-2 Whole Genome Amplication and Sequencing for Effective Population-Based Surveillance and Control of Viral Transmission. In: Clinical Chemistry 66.11 (2020), S. 1450-1458. DOI: 10.1093/clinchem/hvaa187.
Jalees A. Nasir u. a. A Comparison of Whole Genome Sequencing of SARSCoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. In: Viruses 12.8 (2020), S. 895. DOI: 10.3390/v12080895.
Clinton R. Paden u. a. Rapid, sensitive, full-genome sequencing of severe acute respiratory syndrome coronavirus 2. In: Emerging Infectious Diseases 26.10 (2020), S. 2401-2405. DOI: 10.3201/eid2610.201800.
Sureshnee Pillay u. a. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. In: Genes 11.8 (2020), S. 949. DOI: 10.3390/genes11080949.
Dan Hu u. a. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. In: Emerging Microbes & Infections 7.1 (2018), S. 1-10. DOI: 10.1038/s41426-018-0155-5.
Davaalkham Jagdagsuren u. a. The second molecular epidemiological study of HIV infection in Mongolia between 2010 and 2016. In: Plos One 12.12 (2017). DOI: 10.1371/journal.pone.0189605.
J. A. Saldanha, H. C. Thomas und J. P. Monjardino. Cloning and sequencing of RNA of hepatitis delta virus isolated from human serum. In: Journal of General Virology 71.7 (1990), S. 1603-1606. DOI: 10.1099/0022-1317-71-7-1603.
Jernej Mlakar u. a. Zika Virus Associated with Microcephaly. In: New England Journal of Medicine 374.10 (2016), S. 951-958. DOI: 10.1056 /nejmoa1600651.
Christopher L. Parks u. a. Comparison of Predicted Amino Acid Sequences of Measles Virus Strains in the Edmonston Vaccine Lineage. In: Journal of Virology 75.2 (2001), S. 910-920. DOI: 10.1128/jvi.75.2.910-920.2001.
Konstantin M. J. Sparrer u. a. Complete Genome Sequence of a Wild-Type Measles Virus Isolated during the Spring 2013 Epidemic in Germany. In: Genome Announcements 2.2 (2014). DOI: 10.1128/genomea.00157-14.
13. Paul A. Rota u. a. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. In: Science 300.5624 (2003), S. 1394- 1399. DOI: 10.1126/science.1085952.
Runtao He u. a. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. In: Biochemical and Biophysical Research Communications 316.2 (2004), S. 476-483. DOI: 10.1016/j.bbrc.2004.02.074.
Tracey Goldstein u. a. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. In: Nature Microbiology 3.10 (2018), S. 1084- 1089. DOI: 10.1038/s41564-018-0227-2.
Jonathan S. Towner u. a. Marburgvirus Genomics and Association with a Large Hemorrhagic Fever Outbreak in Angola. In: Journal of Virology 80.13 (2006), S. 6497-6516. DOI: 10.1128/jvi.00069-06.
Annika Brinkmann u. a. Amplicov: Rapid whole-genome sequencing using multiplex PCR amplication and real-time Oxford Nanopore minion sequencing enables rapid variant identication of SARS-COV-2. In: Frontiers in Microbiology 12 (2021). DOI: 10.3389/fmicb.2021.651151.
SARS-COV-2. url: https://artic.network/ncov-2019.
Ncbi. ncbi/sra-tools: SRA Tools. URL: https://github.com/ncbi/sra-tools.
[20a] Dinghua Li u. a. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. In: Bioinformatics 31.10 (2015), S. 1674-1676. DOI: 10.1093/bioinformatics/btv033.
[20b] Voutcn. voutcn/megahit: Ultra-fast and memory-ecient (meta-)genome assembler. URL: https://github.com/voutcn/megahit.
[21a] Shifu Chen u. a. fastp: an ultra-fast all-in-one FASTQ preprocessor. In:
Bioinformatics 34.17 (2018), S. i884-i890. DOI: 10.1093/bioinformatics/bty560.
[21b] OpenGene. OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/trimming/ltering/splitting/merging...) URL:
https://github.com/OpenGene/fastp.
[22a] Ben Langmead u. a. Scaling read aligners to hundreds of threads on generalpurpose processors. In: Bioinformatics 35.3 (2018), S. 421-432. DOI:
10. 1093/bioinformatics/bty648.
[22b] Ben Langmead. BenLangmead/bowtie2: A fast and sensitive gapped read aligner. URL: https://github.com/BenLangmead/bowtie2.
[23a] Brian Bushnell. BBMap: A Fast, Accurate, Splice-Aware Aligner. In: (March 2014). URL: https://www.osti.gov/biblio/1241166.
[23b] BBMap. url: https://sourceforge.net/projects/bbmap/.
[24a] Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. In: (May 2013). URL: https://arxiv.org/abs/1303.3997.
[24b] lh3. lh3/bwa: Burrow-Wheeler Aligner for short-read alignment (see mini-map2 for long-read alignment). URL: https://github.com/lh3/bwa.
[25a] H. Li u. a. The Sequence Alignment/Map format and SAMtools. In: Bioinformatics 25.16 (2009), S. 2078-2079. DOI: 10.1093/bioinformatics/btp352.
[25b] Samtools. url:
http://www.htslib.org/
[25c] P. Danecek u. a. Twelve years of SAMtools and BCFtools. In: GigaScience 10.2 (2021). DOI: 10.1093/gigascience/giab008.
[25d] P. Danecek u. a. The variant call format and VCFtools". In: Bioinformatics 27.15 (2011), S. 2156-2158. DOI: 10.1093/bioinformatics/btr330.
[26] Tablet. URL: https://ics.hutton.ac.uk/tablet/.
[27a] Wei Shen u. a. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. In: Plos One 11.10 (2016). DOI: 10.1371/journal.pone.0163962.
[27b] lh3. lh3/seqtk: Toolkit for processing sequences in FASTA/Q formats. URL: https://github.com/lh3/seqtk.
[28] Albrecht Irle. Wahrscheinlichkeitstheorie und Statistik: Grundlagen - Resultate - Anwendungen. Teubner, 2010.
Αν σας άρεσε αυτό το άρθρο, μοιραστείτε το, εγγραφείτε για να λαμβάνετε περισσότερο περιεχόμενο και αν θέλετε να στηρίξετε το συνεχές έργο μου, μπορείτε να χρησιμοποιήσετε τον παρακάτω σύνδεσμο.


—Δικτυογραφία:
SARS-CoV-2 Fragments of Fiction: How Mathematical Fraud and Genetic Sequencing Manufactured a Global Crisis


